April 2nd, 2015
Installation
This is a short introduction on how to use BiocParallel (Morgan, Lang, and Thompson, 2015) and knitrBootstrap (Hester, 2014).
You will need R 3.1.2 or newer (available from CRAN) and BiocParallel
## Install BiocParallel
source('http://bioconductor.org/biocLite.R')
biocLite('BiocParallel')
You will also need knitrBootstrap
## If needed:
# install.packages('devtools')
devtools::install_github('jimhester/knitrBootstrap')
Docs
You can find the latest documentation at
- Introduction to BiocParallel vignette
- knitrBootstrap README
- help pages
help(package = 'BiocParallel') help(package = 'knitrBootstrap')
Parallel computing?
Hm…


plot(y = 10 / (1:10), 1:10, xlab = 'Number of cores', ylab = 'Time',
main = 'Ideal scenario', type = 'o', col = 'blue',
cex = 2, cex.axis = 2, cex.lab = 1.5, cex.main = 2, pch = 16)

plot(y = 10 / (1:10), 1:10, xlab = 'Number of cores', ylab = 'Time',
main = 'Reality', type = 'o', col = 'blue',
cex = 2, cex.axis = 2, cex.lab = 1.5, cex.main = 2, pch = 16)
lines(y = 10 / (1:10) * c(1, 1.05^(2:10) ), 1:10, col = 'red',
type = 'o', cex = 2)

CRAN Task View: High-Performance and Parallel Computing with R

BiocParallel authors



Birthday example
birthday <- function(n) {
m <- 10000
x <- numeric(m)
for(i in 1:m) {
b <- sample(1:365, n, replace = TRUE)
x[i] <- ifelse(length(unique(b)) == n, 0, 1)
}
mean(x)
}
system.time( lapply(1:100, birthday) )
## user system elapsed ## 24.119 0.251 24.388
Source slide 24
Via doMC
library('doMC')
## Loading required package: foreach ## foreach: simple, scalable parallel programming from Revolution Analytics ## Use Revolution R for scalability, fault tolerance and more. ## http://www.revolutionanalytics.com ## Loading required package: iterators ## Loading required package: parallel
registerDoMC(2) system.time( x <- foreach(j = 1:100) %dopar% birthday(j) )
## user system elapsed ## 19.263 0.315 22.145
- Have to change code
- Want to try another parallel mode? Change code again
Via BiocParallel
library('BiocParallel')
system.time( y <- bplapply(1:100, birthday) )
## user system elapsed ## 0.238 0.075 12.789
- Very easy: change lapply() to
bplapply() - Can set global options
Registered modes
registered()
## $MulticoreParam ## class: MulticoreParam ## bpworkers: 4 ## bpisup: FALSE ## bplog: FALSE ## bpthreshold: ## bplogdir: ## bpresultdir: ## bpstopOnError: FALSE ## cluster type: FORK ## ## $SnowParam ## class: SnowParam ## bpworkers: 4 ## bpisup: FALSE ## bplog: FALSE ## bpthreshold: INFO ## bplogdir: ## bpresultdir: ## bpstopOnError: FALSE ## cluster type: SOCK ## ## $SerialParam ## class: SerialParam ## bpworkers: 1
Change modes
## Test in serial mode
system.time( y.serial <- bplapply(1:10, birthday,
BPPARAM = SerialParam()) )
## user system elapsed ## 2.103 0.017 2.124
## Try Snow
system.time( y.snow <- bplapply(1:10, birthday,
BPPARAM = SnowParam(workers = 2)) )
## user system elapsed ## 0.026 0.019 1.738
Use in JHPCE
$ R
> library('BiocParallel')
> registered()
$MulticoreParam
class: MulticoreParam; bpisup: TRUE; bpworkers: 8; catch.errors: TRUE
setSeed: TRUE; recursive: TRUE; cleanup: TRUE; cleanupSignal: 15;
verbose: FALSE
$SnowParam
class: SnowParam; bpisup: FALSE; bpworkers: 8; catch.errors: TRUE
cluster spec: 8; type: PSOCK
$BatchJobsParam
class: BatchJobsParam; bpisup: TRUE; bpworkers: NA; catch.errors: TRUE
cleanup: TRUE; stop.on.error: FALSE; progressbar: TRUE
$SerialParam
class: SerialParam; bpisup: TRUE; bpworkers: 1; catch.errors: TRUE
BatchJobs
- Submits jobs for you in many cluster environments
- Lots of docs: rdocumentation.org/packages/BatchJobs
- Also https://github.com/tudo-r/BatchJobs/wiki/Configuration
.BatchJobs.R file
cluster.functions = makeClusterFunctionsSGE("~/simple.tmpl")
mail.start = "none"
mail.done = "none"
mail.error = "none"
staged.queries = TRUE
fs.timeout = 10
Via Prasad Patil
simple.tmpl file
#!/bin/bash # Job name #$ -N <%= job.name %> # Use current directory #$ -cwd # Get emails #$ -m e R CMD BATCH --no-save --no-restore "<%= rscript %>" /dev/stdout exit 0
Modified from Prasad's version.
I like emails to then explore stats using Alyssa's efficency SGE analytics: code.
General simple.tmpl file
#!/bin/bash # The name of the job, can be anything, simply used when displaying the list of running jobs #$ -N <%= job.name %> # Combining output/error messages into one file #$ -j y # Giving the name of the output log file #$ -o <%= log.file %> # One needs to tell the queue system to use the current directory as the working directory # Or else the script may fail as it will execute in your top level home directory /home/username #$ -cwd # use environment variables #$ -V # use correct queue #$ -q <%= resources$queue %> # use job arrays #$ -t 1-<%= arrayjobs %> # we merge R output with stdout from SGE, which gets then logged via -o option R CMD BATCH --no-save --no-restore "<%= rscript %>" /dev/stdout exit 0
Actually using it
library('BiocParallel')
library('BatchJobs')
# define birthday() function
## Register cluster
funs <- makeClusterFunctionsSGE("~/simple.tmpl")
param <- BatchJobsParam(workers = 10, resources = list(ncpus = 1),
cluster.functions = funs)
register(param)
## Run
system.time( xx <- bplapply(1:100, birthday) )
## Jobs spend a little bit of time in the queue
# user system elapsed
# 0.597 0.350 31.644
Important note
For developers:
Developers wishing to invoke back-ends other than MulticoreParam need to take special care to ensure that required packages, data, and functions are available and loaded on the remote nodes.
Source: BiocParallel vignette
Why should I use BiocParallel?
- Simple to use
- Try different parallel backends without changing code
- Can use it to submit cluster jobs
- Great support from Bioconductor team
HTML reports
People like them because
- easy to share
- we all use the web
- can have interactive features
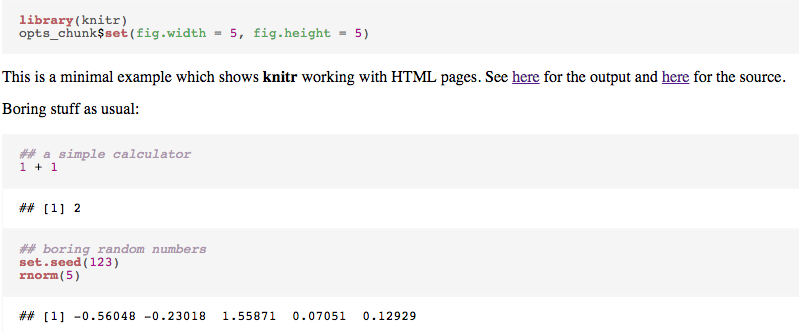
rmarkdown(Allaire, McPherson, Xie, Wickham, et al., 2014) has simplified the process- http://rmarkdown.rstudio.com/
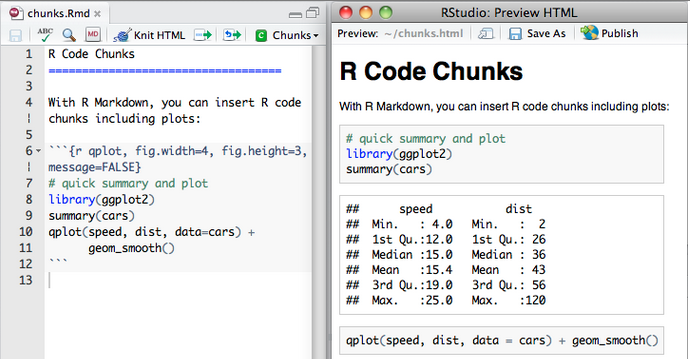
knitrBootstrap
Creating awesome reports for multiple audiences using knitrBootstrap
- Idea:
- One document with code and output
- Hide code by default
- Users who like code, can easily look at it
- Users who don't need code don't get confused by it
Example myFile.Rmd
---
output:
knitrBootstrap::bootstrap_document:
theme.chooser: TRUE
highlight.chooser: TRUE
---
Title
====
Etc
Then render:
rmarkdown::render('myFile.Rmd')
Useful chunk options
bootstrap.show.code = FALSE bootstrap.show.warning = FALSE bootstrap.show.message = FALSE
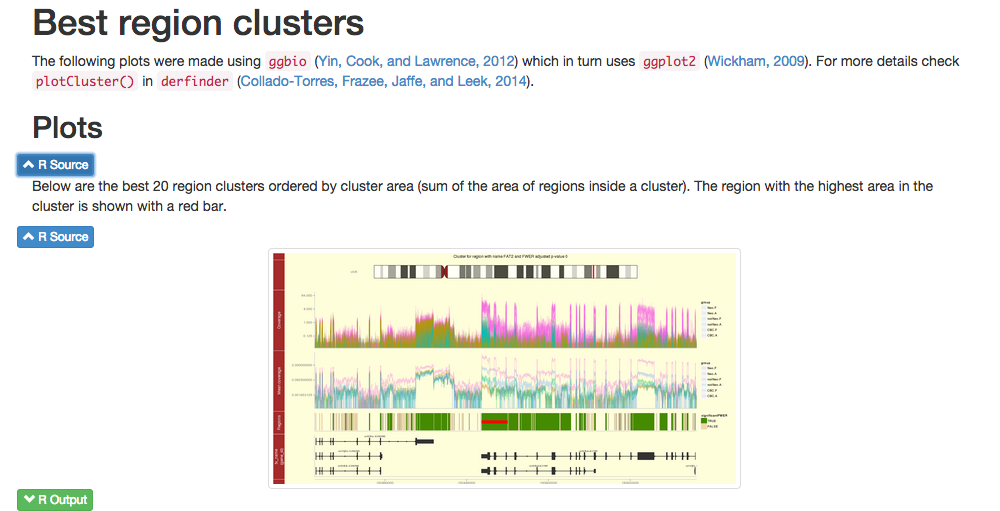
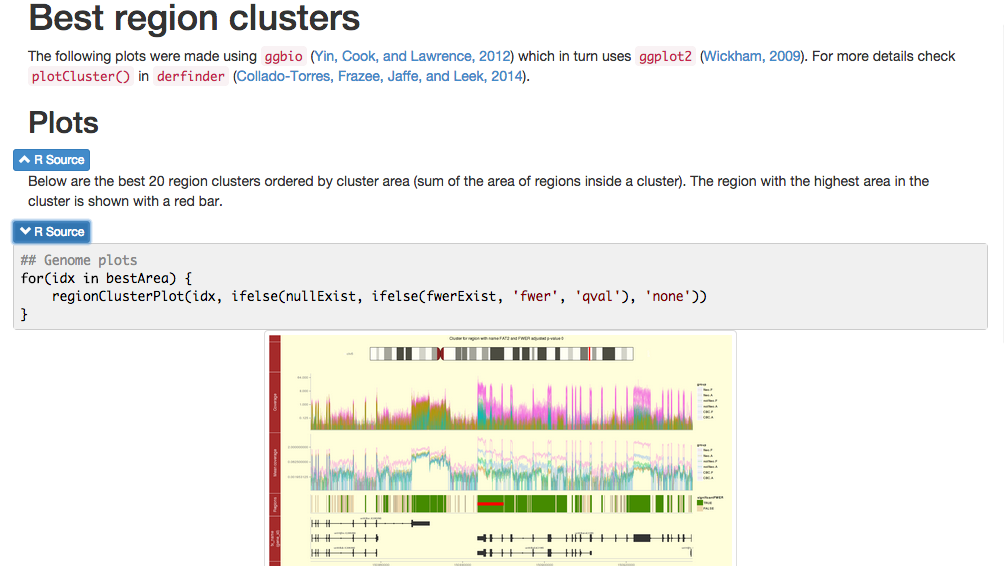
Need help?
- Post in the Bioconductor support site – remember to use the
BiocParalleltag! - For
knitrBootstrap, open a new issue in the GitHub repo
Citing them
## Citation info
citation('BiocParallel')
## Warning in citation("BiocParallel"): no date field in DESCRIPTION file of
## package 'BiocParallel'
##
## To cite package 'BiocParallel' in publications use:
##
## Martin Morgan, Michel Lang and Ryan Thompson (). BiocParallel:
## Bioconductor facilities for parallel evaluation. R package
## version 1.1.21.
##
## A BibTeX entry for LaTeX users is
##
## @Manual{,
## title = {BiocParallel: Bioconductor facilities for parallel evaluation},
## author = {Martin Morgan and Michel Lang and Ryan Thompson},
## note = {R package version 1.1.21},
## }
citation('knitrBootstrap')
##
## To cite package 'knitrBootstrap' in publications use:
##
## Jim Hester (2014). knitrBootstrap: Knitr Bootstrap framework.. R
## package version 1.0.0. https://github.com/jimhester/
##
## A BibTeX entry for LaTeX users is
##
## @Manual{,
## title = {knitrBootstrap: Knitr Bootstrap framework.},
## author = {Jim Hester},
## year = {2014},
## note = {R package version 1.0.0},
## url = {https://github.com/jimhester/},
## }
##
## ATTENTION: This citation information has been auto-generated from
## the package DESCRIPTION file and may need manual editing, see
## 'help("citation")'.
Reproducibility
Code for creating this page
## Create this page
library('rmarkdown')
render('index.Rmd')
## Clean up
file.remove('BiocParallel-knitrBootstrap.bib')
## Extract the R code
library('knitr')
knit('index.Rmd', tangle = TRUE)
Date this tutorial was generated.
## [1] "2015-04-02 01:31:11 EDT"
Wallclock time spent running this tutorial.
## Time difference of 1.095 mins
R session information.
## setting value ## version R Under development (unstable) (2014-11-01 r66923) ## system x86_64, darwin10.8.0 ## ui AQUA ## language (EN) ## collate en_US.UTF-8 ## tz America/New_York
## package * version date source ## bibtex 0.4.0 2014-12-31 CRAN (R 3.2.0) ## BiocParallel * 1.1.21 2015-03-24 Bioconductor ## bitops 1.0.6 2013-08-17 CRAN (R 3.2.0) ## codetools 0.2.11 2015-03-10 CRAN (R 3.2.0) ## devtools * 1.6.1 2014-10-07 CRAN (R 3.2.0) ## digest 0.6.8 2014-12-31 CRAN (R 3.2.0) ## doMC * 1.3.3 2014-02-28 CRAN (R 3.2.0) ## evaluate 0.5.5 2014-04-29 CRAN (R 3.2.0) ## foreach * 1.4.2 2014-04-11 CRAN (R 3.2.0) ## formatR 1.0 2014-08-25 CRAN (R 3.2.0) ## futile.logger 1.4 2015-03-21 CRAN (R 3.2.0) ## futile.options 1.0.0 2010-04-06 CRAN (R 3.2.0) ## htmltools 0.2.6 2014-09-08 CRAN (R 3.2.0) ## httr 0.5 2014-09-02 CRAN (R 3.2.0) ## iterators * 1.0.7 2014-04-11 CRAN (R 3.2.0) ## knitcitations * 1.0.4 2014-11-03 Github (cboettig/knitcitations@508de74) ## knitr 1.7 2014-10-13 CRAN (R 3.2.0) ## lambda.r 1.1.7 2015-03-20 CRAN (R 3.2.0) ## lubridate 1.3.3 2013-12-31 CRAN (R 3.2.0) ## memoise 0.2.1 2014-04-22 CRAN (R 3.2.0) ## plyr 1.8.1 2014-02-26 CRAN (R 3.2.0) ## Rcpp 0.11.5 2015-03-06 CRAN (R 3.2.0) ## RCurl 1.95.4.5 2014-12-28 CRAN (R 3.2.0) ## RefManageR 0.8.40 2014-10-29 CRAN (R 3.2.0) ## RJSONIO 1.3.0 2014-07-28 CRAN (R 3.2.0) ## rmarkdown * 0.3.3 2014-09-17 CRAN (R 3.2.0) ## rstudioapi 0.2 2014-12-31 CRAN (R 3.2.0) ## snow 0.3.13 2013-09-27 CRAN (R 3.2.0) ## stringr 0.6.2 2012-12-06 CRAN (R 3.2.0) ## XML 3.98.1.1 2013-06-20 CRAN (R 3.2.0) ## yaml 2.1.13 2014-06-12 CRAN (R 3.2.0)
Bibliography
This tutorial was generated using rmarkdown (Allaire, McPherson, Xie, Wickham, et al., 2014) and knitcitations (Boettiger, 2015).
[1] J. Allaire, J. McPherson, Y. Xie, H. Wickham, et al. rmarkdown: Dynamic Documents for R. R package version 0.3.3. 2014. URL: http://CRAN.R-project.org/package=rmarkdown.
[2] C. Boettiger. knitcitations: Citations for knitr markdown files. R package version 1.0.4. 2015. URL: https://github.com/cboettig/knitcitations.
[3] J. Hester. knitrBootstrap: Knitr Bootstrap framework. R package version 1.0.0. 2014. URL: https://github.com/jimhester/.
[4] M. Morgan, M. Lang and R. Thompson. BiocParallel: Bioconductor facilities for parallel evaluation. R package version 1.1.21. 2015.