Human splicing diversity and the extent of unannotated splice junctions across human RNA-seq samples on the Sequence Read Archive
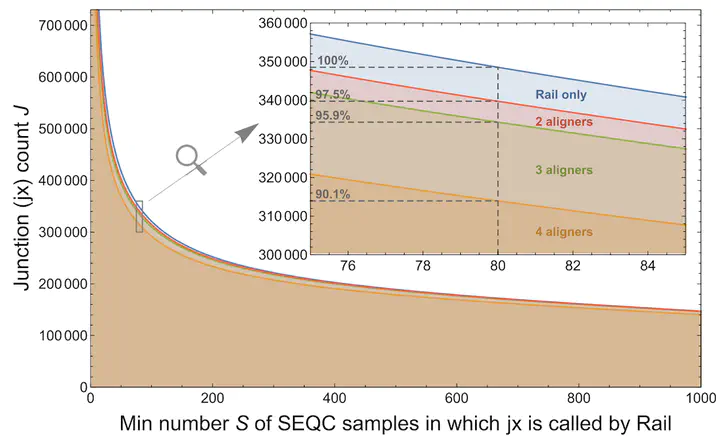 Image credit: bioRxiv
Image credit: bioRxiv
Abstract
Background: Gene annotations, such as those in GENCODE, are derived primarily from alignments of spliced cDNA sequences and protein sequences. The impact of RNA-seq data on annotation has been confined to major projects like ENCODE and Illumina Body Map 2.0. Results: We aligned 21,504 Illumina-sequenced human RNA-seq samples from the Sequence Read Archive (SRA) to the human genome and compared detected exon-exon junctions with junctions in several recent gene annotations. We found 56,861 junctions (18.6%) in at least 1000 samples that were not annotated, and their expression associated with tissue type. Junctions well expressed in individual samples tended to be annotated. Newer samples contributed few novel well-supported junctions, with the vast majority of detected junctions present in samples before 2013. We compiled junction data into a resource called intropolis available at http://intropolis.rail.bio. We used this resource to search for a recently validated isoform of the ALK gene and characterized the potential functional implications of unannotated junctions with publicly available TRAP-seq data. Conclusions: Considering only the variation contained in annotation may suffice if an investigator is interested only in well-expressed transcript isoforms. However, genes that are not generally well expressed and nonetheless present in a small but significant number of samples in the SRA are likelier to be incompletely annotated. The rate at which evidence for novel junctions has been added to the SRA has tapered dramatically, even to the point of an asymptote. Now is perhaps an appropriate time to update incomplete annotations to include splicing present in the now-stable snapshot provided by the SRA.
Human splicing diversity across the Sequence Read Archive https://t.co/wOX5U0Uenm #bioRxiv
— bioRxiv (@biorxivpreprint) January 30, 2016