Improving the value of public RNA-seq expression data by phenotype prediction
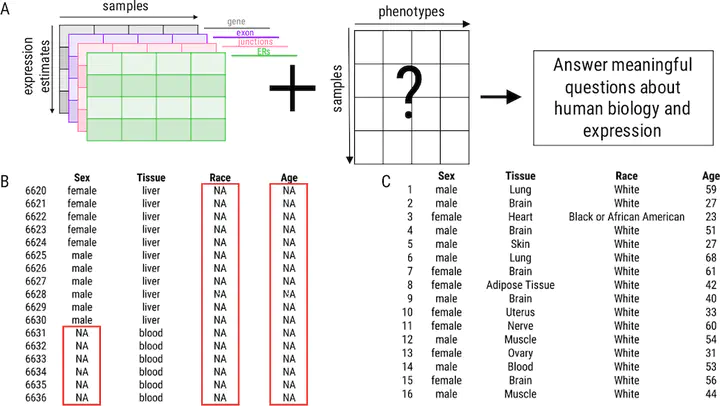 Image credit: NAR
Image credit: NAR
Abstract
Background: Publicly available genomic data are a valuable resource for studying normal human variation and disease, but these data are often not well labeled or annotated. The lack of phenotype information for public genomic data severely limits their utility for addressing targeted biological questions. Results: We develop an in silico phenotyping approach for predicting critical missing annotation directly from genomic measurements using, well-annotated genomic and phenotypic data produced by consortia like TCGA and GTEx as training data. We apply in silico phenotyping to a set of 70,000 RNA-seq samples we recently processed on a common pipeline as part of the recount2 project (https://jhubiostatistics.shinyapps.io/recount/). We use gene expression data to build and evaluate predictors for both biological phenotypes (sex, tissue, sample source) and experimental conditions (sequencing strategy). We demonstrate how these predictions can be used to study cross-sample properties of public genomic data, select genomic projects with specific characteristics, and perform downstream analyses using predicted phenotypes. The methods to perform phenotype prediction are available in the phenopredict R package (https://github.com/leekgroup/phenopredict) and the predictions for recount2 are available from the recount R package (https://bioconductor.org/packages/release/bioc/html/recount.html). Conclusion: Having leveraging massive public data sets to generate a well-phenotyped set of expression data for more than 70,000 human samples, expression data is available for use on a scale that was not previously feasible.
Congrats to @Shannon_E_Ellis on her work improving the value of public genomic data by phenotype prediction now out at @NAR_Open: https://t.co/SWQvTuKMxk you can get the predictions from the recount package: https://t.co/y8CDPephE4 pic.twitter.com/jjTQIePKet
— Jeff Leek (@jtleek) March 5, 2018
Access all of @Shannon_E_Ellis’s predictions by using recount::add_predictions() [with no rse argument] in recount version >= 1.5.9. Great work Shannon! #rstats #rnaseq #bioinformatics https://t.co/g0yt90AzSp
— 🇲🇽 Leonardo Collado-Torres (@lcolladotor) March 6, 2018
Leonardo Collado-Torres
Investigator @ LIBD, Assistant Professor, Department of Biostatistics @ JHBSPH
#rstats @Bioconductor/🧠 genomics @LieberInstitute/@lcgunam @jhubiostat @jtleek @andrewejaffe alumni/@LIBDrstats @CDSBMexico co-founder