Challenges and opportunities to computationally deconvolve heterogeneous tissue with varying cell sizes using single-cell RNA-sequencing datasets
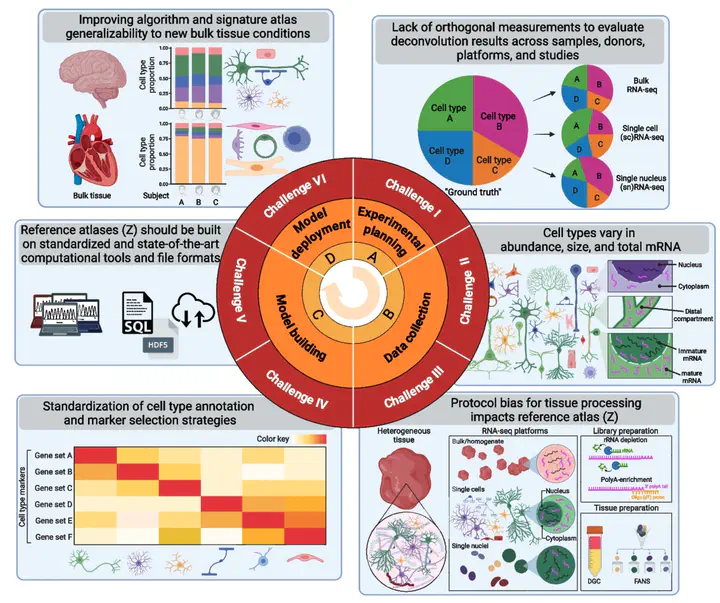 Image credit: arRxiv
Image credit: arRxiv
Abstract
Deconvolution of cell mixtures in “bulk” transcriptomic samples from homogenate human tissue is important for understanding disease pathologies. However, several experimental and computational challenges impede transcriptomics-based deconvolution approaches using single-cell/nucleus RNA-seq reference atlases. Cells from the brain and blood have substantially different sizes, total mRNA, and transcriptional activities, and existing approaches may quantify total mRNA instead of cell type proportions. Further, standards are lacking for the use of cell reference atlases and integrative analyses of single-cell and spatial transcriptomics data. We discuss how to approach these key challenges with orthogonal “gold standard” datasets for evaluating deconvolution methods.
You can now read my first preprint as a @jhubiostat postdoc, in collaboration with @LieberInstitute -- a commentary on #transcriptomics #deconvolution challenges & opportunities for heterogeneous tissues! A tweetorial... 1/ 🧬🧪💻🔥#deconvochallenge 🔥 https://t.co/GsvpMgTTPj
— Sean Maden (@MadenSean) May 15, 2023
Louise A. Huuki-Myers
Research Associate 2020-2022, Staff Scientist I, Data Science 2022-ongoing, PhD Student 2024-ongoing
Leonardo Collado-Torres
Investigator @ LIBD, Assistant Professor, Department of Biostatistics @ JHBSPH
#rstats @Bioconductor/🧠 genomics @LieberInstitute/@lcgunam @jhubiostat @jtleek @andrewejaffe alumni/@LIBDrstats @CDSBMexico co-founder