Detection of pathogenic splicing events from RNA-sequencing data using dasper
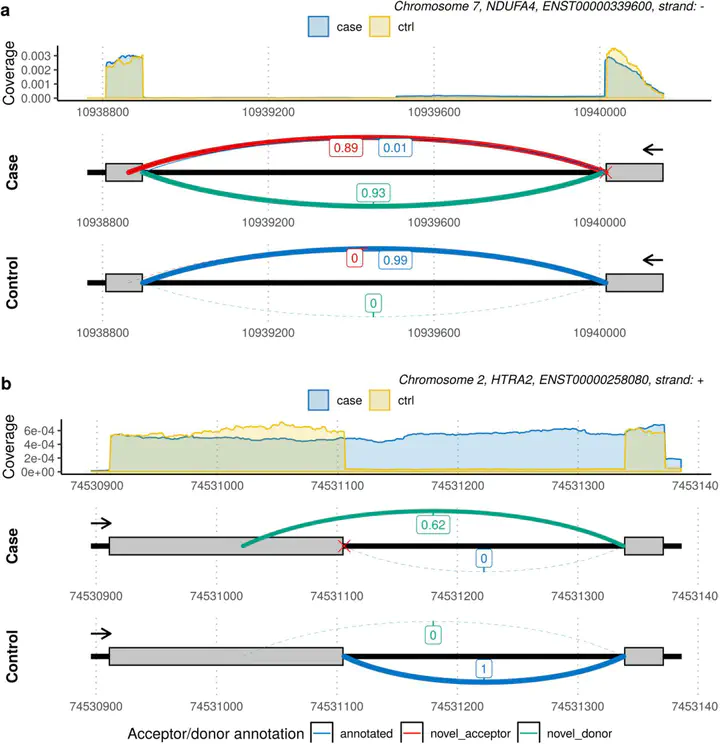 Image credit: bioRxiv
Image credit: bioRxiv
Abstract
Although next-generation sequencing technologies have accelerated the discovery of novel gene-to-disease associations, many patients with suspected Mendelian diseases still leave the clinic without a genetic diagnosis. An estimated one third of these patients will have disorders caused by mutations impacting splicing. RNA-sequencing has been shown to be a promising diagnostic tool, however few methods have been developed to integrate RNA-sequencing data into the diagnostic pipeline. Here, we introduce dasper, an R/Bioconductor package that improves upon existing tools for detecting aberrant splicing by using machine learning to incorporate disruptions in exon-exon junction counts as well as coverage. dasper is designed for diagnostics, providing a rank-based report of how aberrant each splicing event looks, as well as including visualization functionality to facilitate interpretation. We validate dasper using 16 patient-derived fibroblast cell lines harbouring pathogenic variants known to impact splicing. We find that dasper is able to detect pathogenic splicing events with greater accuracy than existing LeafCutterMD or z-score approaches. Furthermore, by only applying a broad OMIM gene filter (without any variant-level filters), dasper is able to detect pathogenic splicing events within the top 10 most aberrant identified for each patient. Since using publicly available control data minimises costs associated with incorporating RNA-sequencing into diagnostic pipelines, we also investigate the use of 504 GTEx fibroblast samples as controls. We find that dasper leverages publicly available data effectively, ranking pathogenic splicing events in the top 25. Thus, we believe dasper can increase diagnostic yield for a pathogenic splicing variants and enable the efficient implementation of RNA-sequencing for diagnostics in clinical laboratories.
This week was very intense but lots of new #rstats 📦 goodies are now under review at @Bioconductor 🤞🏽#biocthis https://t.co/p3vu5DxRsi#recount3 https://t.co/ogg7QFm0gq#megadepth & #dasper https://t.co/AFRs6nVt1a
— 🇲🇽 Leonardo Collado-Torres (@lcolladotor) October 2, 2020
Congrats on your 1st & 2nd 📦 submission @dyzhang32!! 🤩 pic.twitter.com/mMYDEnzUL8
Leonardo Collado-Torres
Investigator @ LIBD, Assistant Professor, Department of Biostatistics @ JHBSPH
#rstats @Bioconductor/🧠 genomics @LieberInstitute/@lcgunam @jhubiostat @jtleek @andrewejaffe alumni/@LIBDrstats @CDSBMexico co-founder