RNA-seq transcript quantification from reduced-representation data in recount2
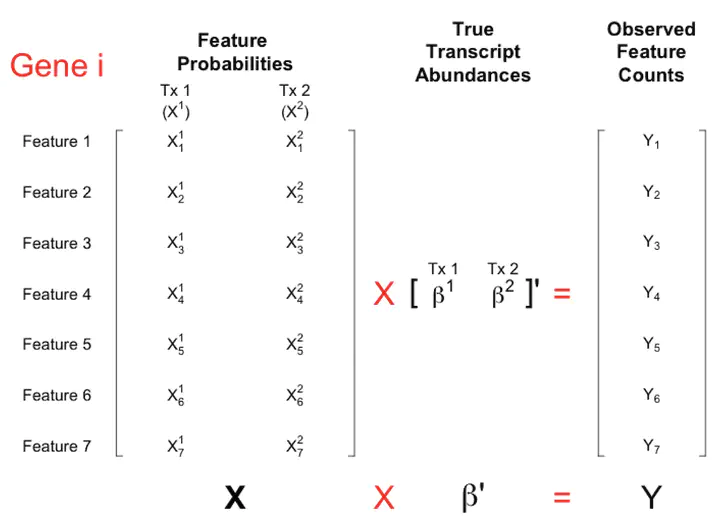 Image credit: bioRxiv
Image credit: bioRxiv
Abstract
More than 70,000 short-read RNA-sequencing samples are publicly available through the recount2 project, a curated database of summary coverage data. However, no current methods can estimate transcript-level abundances using the reduced-representation information stored in this database. Here we present a linear model utilizing coverage of junctions and subdivided exons to generate transcript abundance estimates of comparable accuracy to those obtained from methods requiring read-level data. Our approach flexibly models bias, produces standard errors, and is easy to refresh given updated annotation. We illustrate our method on simulated and real data and release transcript abundance estimates for the samples in recount2.
We now have transcript abundances for recount2! https://t.co/7z6fcJ6qwF
— Jack Fu (@JFuBiostats) January 13, 2018
Leonardo Collado-Torres
Investigator @ LIBD, Assistant Professor, Department of Biostatistics @ JHBSPH
#rstats @Bioconductor/🧠 genomics @LieberInstitute/@lcgunam @jhubiostat @jtleek @andrewejaffe alumni/@LIBDrstats @CDSBMexico co-founder