Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer's disease
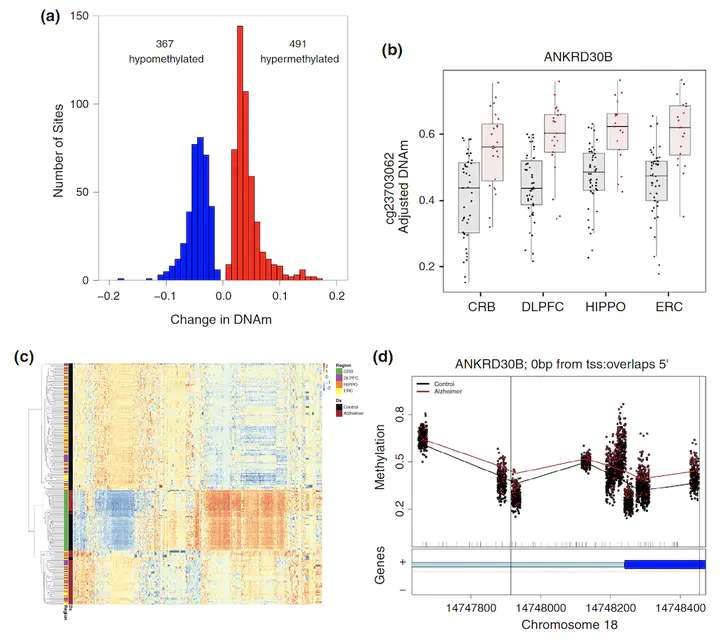 Image credit: Acta Neuropathologica
Image credit: Acta Neuropathologica
Abstract
Late-onset Alzheimer’s disease (AD) is a complex age-related neurodegenerative disorder that likely involves epigenetic factors. To better understand the epigenetic state associated with AD, we surveyed 420,852 DNA methylation (DNAm) sites from neurotypical controls (N=49) and late-onset AD patients (N=24) across four brain regions (hippocampus, entorhinal cortex, dorsolateral prefrontal cortex and cerebellum). We identified 858 sites with robust differential methylation collectively annotated to 772 possible genes (FDR<5%, within 10 kb). These sites were overrepresented in AD genetic risk loci (p=0.00655) and were enriched for changes during normal aging (p<2.2×10−16), and nearby genes were enriched for processes related to cell-adhesion, immunity, and calcium homeostasis (FDR<5%). To functionally validate these associations, we generated and analyzed corresponding transcriptome data to prioritize 130 genes within 10 kb of the differentially methylated sites. These 130 genes were differentially expressed between AD cases and controls and their expression was associated with nearby DNAm (p<0.05). This integrated analysis implicates novel genes in Alzheimer’s disease, such as ANKRD30B. These results highlight DNAm differences in Alzheimer’s disease that have gene expression correlates, further implicating DNAm as an epigenetic mechanism underlying pathological molecular changes associated with AD. Furthermore, our framework illustrates the value of integrating epigenetic and transcriptomic data for understanding complex disease.
Fresh off the press: Integrated DNA methylation and gene expression profling across multiple brain regions implicate novel genes in Alzheimer’s diseasehttps://t.co/a35BM6decX
— 🇲🇽 Leonardo Collado-Torres (@lcolladotor) February 4, 2019
Lead by @SteveSemick with all @LieberInstitute co-authors GEO: GSE125895 Code: https://t.co/UoXVFShSo6 pic.twitter.com/5DbfEkq0Fo
Leonardo Collado-Torres
Investigator @ LIBD, Assistant Professor, Department of Biostatistics @ JHBSPH
#rstats @Bioconductor/🧠 genomics @LieberInstitute/@lcgunam @jhubiostat @jtleek @andrewejaffe alumni/@LIBDrstats @CDSBMexico co-founder