4 Datos de RNA-seq a través de recount3
4.1 Procesar datos crudos (FASTQ) con SPEAQeasy
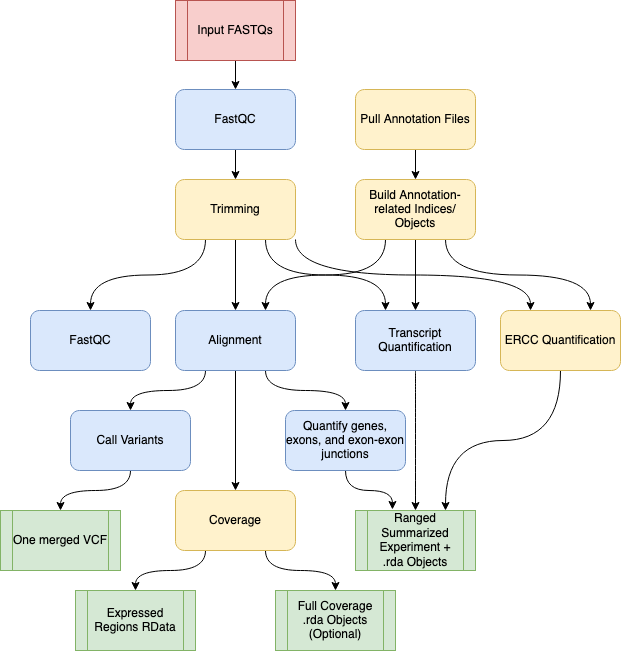
- Pre-print de diciembre 2020 https://www.biorxiv.org/content/10.1101/2020.12.11.386789v1
- Artículo de mayo 2021 https://doi.org/10.1186/s12859-021-04142-3
- Documentación del software http://research.libd.org/SPEAQeasy/
- Ejemplo de como usar el software y analizar los datos http://research.libd.org/SPEAQeasy-example/
- Taller de 3 días (aprox 6 horas en total) sobre expresión diferencial usando estos datos https://lcolladotor.github.io/bioc_team_ds/differential-expression-analysis.html
4.2 Proyectos recount
ReCount: datos de unos 20 estudiosrecount: 70 mil muestras de RNA-seq uniformemente procesadas- https://jhubiostatistics.shinyapps.io/recount/
- Documentación con
pkgdown: http://leekgroup.github.io/recount/ - Documentación en Bioconductor: http://bioconductor.org/packages/recount
- Artículo principal de 2017 http://www.nature.com/nbt/journal/v35/n4/full/nbt.3838.html
- Artículo que explica porque las cuentas (counts) son diferentes a las usuales https://f1000research.com/articles/6-1558/v1
- Algunos análisis ejemplo que hicimos http://leekgroup.github.io/recount-analyses/
recount3: 700 mil muestras de RNA-seq de humano y ratón- http://rna.recount.bio/
- Documentación con
pkgdown: http://research.libd.org/recount3/ - Documentación en Bioconductor: http://bioconductor.org/packages/recount3
- Pre-print: mayo 2021 https://doi.org/10.1101/2021.05.21.445138
- Artículo: noviembre 2021 https://doi.org/10.1186/s13059-021-02533-6
- Ayuda a que todxs podamos analizar los datos sin importar quien tiene acceso a high performance computing (HPC) (clústers para procesar datos).
- Es como democratizar el acceso a los datos ^^
4.3 Usar recount3
Check the original documentation in English here and here.
Primero cargamos el paquete de R que automáticamente carga todas las dependencias incluyendo a SummarizedExperiment.
## Load recount3 R package
library("recount3")Después tenemos que identificar un estudio de interes y determinar si queremos accesar la información a nivel de genes, exones, etc. Sabiendo el estudio de interes, podemos descargar los datos usando la función create_rse() como mostramos a continuación. create_rse() tiene argumentos con los cuales podemos especificar la anotación que queremos usar (las opciones dependen del organismo).
## Revisemos todos los proyectos con datos de humano en recount3
human_projects <- available_projects()## 2022-02-04 17:18:36 caching file sra.recount_project.MD.gz.## adding rname 'http://duffel.rail.bio/recount3/human/data_sources/sra/metadata/sra.recount_project.MD.gz'## 2022-02-04 17:18:41 caching file gtex.recount_project.MD.gz.## adding rname 'http://duffel.rail.bio/recount3/human/data_sources/gtex/metadata/gtex.recount_project.MD.gz'## 2022-02-04 17:18:43 caching file tcga.recount_project.MD.gz.## adding rname 'http://duffel.rail.bio/recount3/human/data_sources/tcga/metadata/tcga.recount_project.MD.gz'## Encuentra tu proyecto de interés. Aquí usaremos
## SRP009615 de ejemplo
proj_info <- subset(
human_projects,
project == "SRP009615" & project_type == "data_sources"
)
## Crea un objetio de tipo RangedSummarizedExperiment (RSE)
## con la información a nivel de genes
rse_gene_SRP009615 <- create_rse(proj_info)## 2022-02-04 17:18:51 downloading and reading the metadata.## 2022-02-04 17:18:51 caching file sra.sra.SRP009615.MD.gz.## adding rname 'http://duffel.rail.bio/recount3/human/data_sources/sra/metadata/15/SRP009615/sra.sra.SRP009615.MD.gz'## 2022-02-04 17:18:52 caching file sra.recount_project.SRP009615.MD.gz.## adding rname 'http://duffel.rail.bio/recount3/human/data_sources/sra/metadata/15/SRP009615/sra.recount_project.SRP009615.MD.gz'## 2022-02-04 17:18:53 caching file sra.recount_qc.SRP009615.MD.gz.## adding rname 'http://duffel.rail.bio/recount3/human/data_sources/sra/metadata/15/SRP009615/sra.recount_qc.SRP009615.MD.gz'## 2022-02-04 17:18:54 caching file sra.recount_seq_qc.SRP009615.MD.gz.## adding rname 'http://duffel.rail.bio/recount3/human/data_sources/sra/metadata/15/SRP009615/sra.recount_seq_qc.SRP009615.MD.gz'## 2022-02-04 17:18:55 caching file sra.recount_pred.SRP009615.MD.gz.## adding rname 'http://duffel.rail.bio/recount3/human/data_sources/sra/metadata/15/SRP009615/sra.recount_pred.SRP009615.MD.gz'## 2022-02-04 17:18:56 downloading and reading the feature information.## 2022-02-04 17:18:56 caching file human.gene_sums.G026.gtf.gz.## adding rname 'http://duffel.rail.bio/recount3/human/annotations/gene_sums/human.gene_sums.G026.gtf.gz'## 2022-02-04 17:18:58 downloading and reading the counts: 12 samples across 63856 features.## 2022-02-04 17:18:58 caching file sra.gene_sums.SRP009615.G026.gz.## adding rname 'http://duffel.rail.bio/recount3/human/data_sources/sra/gene_sums/15/SRP009615/sra.gene_sums.SRP009615.G026.gz'## 2022-02-04 17:18:59 construcing the RangedSummarizedExperiment (rse) object.## Explora el objeto RSE
rse_gene_SRP009615## class: RangedSummarizedExperiment
## dim: 63856 12
## metadata(8): time_created recount3_version ... annotation recount3_url
## assays(1): raw_counts
## rownames(63856): ENSG00000278704.1 ENSG00000277400.1 ... ENSG00000182484.15_PAR_Y ENSG00000227159.8_PAR_Y
## rowData names(10): source type ... havana_gene tag
## colnames(12): SRR387777 SRR387778 ... SRR389077 SRR389078
## colData names(175): rail_id external_id ... recount_pred.curated.cell_line BigWigURLDe forma interactiva también podemos escoger nuestro estudio de interés usando el siguiente código o vía el explorar de estudios que creamos.
## Explora los proyectos disponibles de forma interactiva
proj_info_interactive <- interactiveDisplayBase::display(human_projects)
## Selecciona un solo renglón en la tabla y da click en "send".
## Aquí verificamos que solo seleccionaste un solo renglón.
stopifnot(nrow(proj_info_interactive) == 1)
## Crea el objeto RSE
rse_gene_interactive <- create_rse(proj_info_interactive)Una vez que tenemos las cuentas, podemos usar transform_counts() o compute_read_counts() para convertir en los formatos esperados por otras herramientas. Revisen el artículo de 2017 del recountWorkflow para más detalles.
## Convirtamos las cuentas por nucleotido a cuentas por lectura
## usando compute_read_counts().
## Para otras transformaciones como RPKM y TPM, revisa transform_counts().
assay(rse_gene_SRP009615, "counts") <- compute_read_counts(rse_gene_SRP009615)## Para este estudio en específico, hagamos más fácil de usar la
## información del experimento
rse_gene_SRP009615 <- expand_sra_attributes(rse_gene_SRP009615)
colData(rse_gene_SRP009615)[
,
grepl("^sra_attribute", colnames(colData(rse_gene_SRP009615)))
]## DataFrame with 12 rows and 4 columns
## sra_attribute.cells sra_attribute.shRNA_expression sra_attribute.source_name sra_attribute.treatment
## <character> <character> <character> <character>
## SRR387777 K562 no SL2933 Puromycin
## SRR387778 K562 yes, targeting SRF SL2934 Puromycin, doxycycline
## SRR387779 K562 no SL5265 Puromycin
## SRR387780 K562 yes targeting SRF SL3141 Puromycin, doxycycline
## SRR389079 K562 no shRNA expression SL6485 Puromycin
## ... ... ... ... ...
## SRR389082 K562 expressing shRNA tar.. SL2592 Puromycin, doxycycline
## SRR389083 K562 no shRNA expression SL4337 Puromycin
## SRR389084 K562 expressing shRNA tar.. SL4326 Puromycin, doxycycline
## SRR389077 K562 no shRNA expression SL1584 Puromycin
## SRR389078 K562 expressing shRNA tar.. SL1583 Puromycin, doxycyclineAhora estamos listos para usar otras herramientas para el análisis de los datos.
4.4 Ejercicio
- Utiliza
iSEEpara reproducir la siguiente imagen
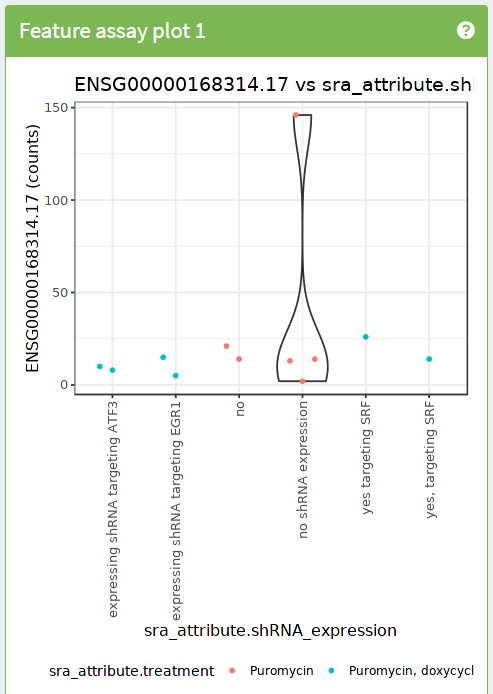
- Pistas:
- Utiliza el dynamic feature selection
- Utiliza información de las columnas para el eje X
- Utiliza información de las columnas para los colores
- (opcional) Crea tu cuenta gratis de https://www.shinyapps.io/ y comparte tu visualización de los datos usando
iSEEde esa forma. Ejemplos reales: https://github.com/LieberInstitute/10xPilot_snRNAseq-human#explore-the-data-interactively.
4.5 Comunidad
- Autores de recount2 y 3 en Twitter:
- Más sobre los tipos de cuentas:
If I'm using recount2 data for a differential analysis in DEseq2, should I be using the original counts, or the scaled counts?@mikelove @lcolladotor #rstats #Bioconductor
— Dr. Robert M Flight, PhD (@rmflight) January 29, 2021
- Ejemplos de tweets de la comunidad
Un tweet de un alumno del 2021:
@lcolladotor
— Axel Zagal Norman (@NormanZagal) February 25, 2021
Earlier I was looking for some data to analyze in recount, they have so much, I seriously can't decide what to use! https://t.co/fIJwXq46Tz
Thanks for such an useful package!@chrisnwilks @BenLangmead @KasperDHansen @AbhiNellore @Shannon_E_Ellis @jtleek
Explorando la posibilidad de hacer análisis con recount3 (enero 2022):
I have found a novel exon expressed in a cancer sample. I would like to search TCGA/SRA to identify other samples with the same/similar exon. It will be rare. Can I use Recount3, megadepth for this? @jtleek @lcolladotor @BenLangmead
— Alicia Oshlack (@AliciaOshlack) January 5, 2022
Un tweet de ayer (febrero 1, 2022):
Thinking on this a bit it is strange how few people are doing “medium-sized” meta analyses of transcriptiomics. One on end you have @BenLangmead @lcolladotor reprocessing (with a touch of analysis) most of SRA. And you see papers pulling an dataset or two to corroborate.
— David McGaughey (@David_McGaughey) February 1, 2022