SPEAQeasy: a scalable pipeline for expression analysis and quantification for R/Bioconductor-powered RNA-seq analyses
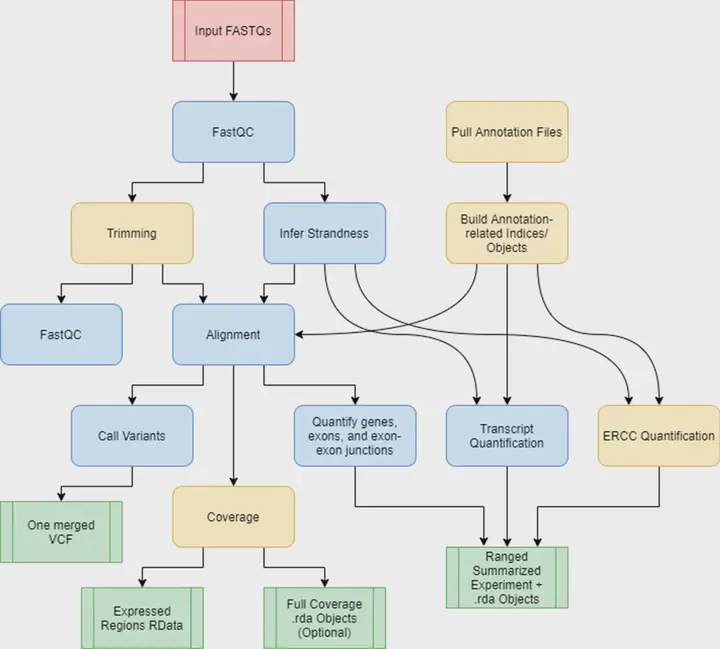 Image credit: BMC Bioinformatics
Image credit: BMC Bioinformatics
Abstract
Background. RNA sequencing (RNA-seq) is a common and widespread biological assay, and an increasing amount of data is generated with it. In practice, there are a large number of individual steps a researcher must perform before raw RNA-seq reads yield directly valuable information, such as differential gene expression data. Existing software tools are typically specialized, only performing one step–such as alignment of reads to a reference genome–of a larger workflow. The demand for a more comprehensive and reproducible workflow has led to the production of a number of publicly available RNA-seq pipelines. However, we have found that most require computational expertise to set up or share among several users, are not actively maintained, or lack features we have found to be important in our own analyses. Results. In response to these concerns, we have developed a Scalable Pipeline for Expression Analysis and Quantification (SPEAQeasy), which is easy to install and share, and provides a bridge towards R/Bioconductor downstream analysis solutions. SPEAQeasy is portable across computational frameworks (SGE, SLURM, local, docker integration) and different configuration files are provided (http://research.libd.org/SPEAQeasy/). Conclusions. SPEAQeasy is user-friendly and lowers the computational-domain entry barrier for biologists and clinicians to RNA-seq data processing as the main input file is a table with sample names and their corresponding FASTQ files. The goal is to provide a flexible pipeline that is immediately usable by researchers, regardless of their technical background or computing environment.
Congrats Nick https://t.co/O3u5XRPXy2 for your @biorxivpreprint first pre-print! 🙌🏽
— 🇲🇽 Leonardo Collado-Torres (@lcolladotor) December 12, 2020
SPEAQeasy is our @nextflowio implementation of the #RNAseq processing pipeline that produces @Bioconductor-friendly #rstats objects that we use at @LieberInstitute
📜 https://t.co/zKuBRtBCmY pic.twitter.com/F83fXI90eP
Nicholas J. Eagles
Research Assistant 2018-2021, Research Associate I 2021-2024, Research Associate II 2024-ongoing
Joshua M. Stolz
Research Associate 2018-2022
Louise A. Huuki-Myers
Research Associate 2020-2022, Staff Scientist I, Data Science 2022-ongoing, PhD Student 2024-ongoing
Leonardo Collado-Torres
Investigator @ LIBD, Assistant Professor, Department of Biostatistics @ JHBSPH
#rstats @Bioconductor/🧠 genomics @LieberInstitute/@lcgunam @jhubiostat @jtleek @andrewejaffe alumni/@LIBDrstats @CDSBMexico co-founder