derfinder quick start guide
Leonardo Collado-Torres
Lieber Institute for Brain Development, Johns Hopkins Medical CampusCenter for Computational Biology, Johns Hopkins Universitylcolladotor@gmail.com
31 March 2026
Source:vignettes/derfinder-quickstart.Rmd
derfinder-quickstart.RmdBasics
Install derfinder
R is an open-source statistical environment which can be
easily modified to enhance its functionality via packages. derfinder
is a R package available via the Bioconductor
repository for packages. R can be installed on any
operating system from CRAN
after which you can install derfinder
by using the following commands in your R session:
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("derfinder")
## Check that you have a valid Bioconductor installation
BiocManager::valid()Required knowledge
derfinder is based on many other packages and in particular in those that have implemented the infrastructure needed for dealing with RNA-seq data. That is, packages like Rsamtools, GenomicAlignments and rtracklayer that allow you to import the data. A derfinder user is not expected to deal with those packages directly but will need to be familiar with GenomicRanges to understand the results derfinder generates. It might also prove to be highly beneficial to check the BiocParallel package for performing parallel computations.
If you are asking yourself the question “Where do I start using Bioconductor?” you might be interested in this blog post.
Asking for help
As package developers, we try to explain clearly how to use our
packages and in which order to use the functions. But R and
Bioconductor have a steep learning curve so it is critical
to learn where to ask for help. The blog post quoted above mentions some
but we would like to highlight the Bioconductor support site
as the main resource for getting help: remember to use the
derfinder tag and check the older
posts. Other alternatives are available such as creating GitHub
issues and tweeting. However, please note that if you want to receive
help you should adhere to the posting
guidelines. It is particularly critical that you provide a small
reproducible example and your session information so package developers
can track down the source of the error.
We would like to highlight the derfinder user Jessica Hekman. She has used derfinder with non-human data, and in the process of doing so discovered some small bugs or sections of the documentation that were not clear.
Citing derfinder
We hope that derfinder will be useful for your research. Please use the following information to cite the package and the overall approach. Thank you!
## Citation info
citation("derfinder")## To cite package 'derfinder' in publications use:
##
## Collado-Torres L, Nellore A, Frazee AC, Wilks C, Love MI, Langmead B,
## Irizarry RA, Leek JT, Jaffe AE (2017). "Flexible expressed region
## analysis for RNA-seq with derfinder." _Nucl. Acids Res._.
## doi:10.1093/nar/gkw852 <https://doi.org/10.1093/nar/gkw852>.
## <http://nar.oxfordjournals.org/content/early/2016/09/29/nar.gkw852>.
##
## Frazee AC, Sabunciyan S, Hansen KD, Irizarry RA, Leek JT (2014).
## "Differential expression analysis of RNA-seq data at single-base
## resolution." _Biostatistics_, *15 (3)*, 413-426.
## doi:10.1093/biostatistics/kxt053
## <https://doi.org/10.1093/biostatistics/kxt053>.
## <http://biostatistics.oxfordjournals.org/content/15/3/413.long>.
##
## Collado-Torres L, Jaffe AE, Leek JT (2017). _derfinder:
## Annotation-agnostic differential expression analysis of RNA-seq data
## at base-pair resolution via the DER Finder approach_.
## doi:10.18129/B9.bioc.derfinder
## <https://doi.org/10.18129/B9.bioc.derfinder>.
## https://github.com/lcolladotor/derfinder - R package version 1.45.0,
## <http://www.bioconductor.org/packages/derfinder>.
##
## To see these entries in BibTeX format, use 'print(<citation>,
## bibtex=TRUE)', 'toBibtex(.)', or set
## 'options(citation.bibtex.max=999)'.Quick start to using to derfinder
Here is a very quick example of a DER Finder analysis. This analysis is explained in more detail later on in this document.
## Load libraries
library("derfinder")
library("derfinderData")
library("GenomicRanges")
## Determine the files to use and fix the names
files <- rawFiles(system.file("extdata", "AMY", package = "derfinderData"),
samplepatt = "bw", fileterm = NULL
)
names(files) <- gsub(".bw", "", names(files))
## Load the data from disk -- On Windows you have to load data from Bam files
fullCov <- fullCoverage(files = files, chrs = "chr21", verbose = FALSE)
## Get the region matrix of Expressed Regions (ERs)
regionMat <- regionMatrix(fullCov, cutoff = 30, L = 76, verbose = FALSE)
## Get pheno table
pheno <- subset(brainspanPheno, structure_acronym == "AMY")
## Identify which ERs are differentially expressed, that is, find the DERs
library("DESeq2")
## Round matrix
counts <- round(regionMat$chr21$coverageMatrix)
## Round matrix and specify design
dse <- DESeqDataSetFromMatrix(counts, pheno, ~ group + gender)
## Perform DE analysis
dse <- DESeq(dse, test = "LRT", reduced = ~gender, fitType = "local")
## Extract results
mcols(regionMat$chr21$regions) <- c(mcols(regionMat$chr21$regions), results(dse))
## Save info in an object with a shorter name
ers <- regionMat$chr21$regions
ersIntroduction
derfinder is an R package that implements the DER Finder approach (Frazee, Sabunciyan, Hansen, Irizarry, and Leek, 2014) for RNA-seq data. Briefly, this approach is an alternative to feature-counting and transcript assembly. The basic idea is to identify contiguous base-pairs in the genome that present differential expression signal. These base-pairs are grouped into _d_ifferentially _e_xpressed _r_regions (DERs). This approach is annotation-agnostic which is a feature you might be interested in. In particular, derfinder contains functions that allow you to identify DERs via two alternative methods. You can find more details on the full derfinder users guide.
Sample DER Finder analysis
This is a brief overview of what a DER Finder analysis looks like. In particular, here we will be identifying expressed regions (ERs) without relying on annotation. Next, we’ll identify candidate differentially expressed regions (DERs). Finally, we’ll compare the DERs with known annotation features.
We first load the required packages.
## Load libraries
library("derfinder")
library("derfinderData")
library("GenomicRanges")Next, we need to locate the chromosome 21 coverage files for a set of
12 samples. These samples are a small subset from the BrainSpan
Atlas of the Human Brain (BrainSpan, 2011) publicly available data.
The function rawFiles() helps us in locating these
files.
## Determine the files to use and fix the names
files <- rawFiles(system.file("extdata", "AMY", package = "derfinderData"),
samplepatt = "bw", fileterm = NULL
)
names(files) <- gsub(".bw", "", names(files))Next, we can load the full coverage data into memory using the
fullCoverage() function. Note that the BrainSpan
data is already normalized by the total number of mapped reads in each
sample. However, that won’t be the case with most data sets in which
case you might want to use the totalMapped and
targetSize arguments. The function
getTotalMapped() will be helpful to get this
information.
## Load the data from disk
fullCov <- fullCoverage(
files = files, chrs = "chr21", verbose = FALSE,
totalMapped = rep(1, length(files)), targetSize = 1
)Now that we have the data, we can identify expressed regions (ERs) by using a cutoff of 30 on the base-level mean coverage from these 12 samples. Once the regions have been identified, we can calculate a coverage matrix with one row per ER and one column per sample (12 in this case). For doing this calculation we need to know the length of the sequence reads, which in this study were 76 bp long.
## Get the region matrix of Expressed Regions (ERs)
regionMat <- regionMatrix(fullCov, cutoff = 30, L = 76, verbose = FALSE)regionMatrix() returns a list of elements, each with
three pieces of output. The actual ERs are arranged in a
GRanges object named regions.
## regions output
regionMat$chr21$regions## GRanges object with 45 ranges and 6 metadata columns:
## seqnames ranges strand | value area indexStart
## <Rle> <IRanges> <Rle> | <numeric> <numeric> <integer>
## 1 chr21 9827018-9827582 * | 313.6717 177224.53 1
## 2 chr21 15457301-15457438 * | 215.0846 29681.68 566
## 3 chr21 20230140-20230192 * | 38.8325 2058.12 704
## 4 chr21 20230445-20230505 * | 41.3245 2520.80 757
## 5 chr21 27253318-27253543 * | 34.9131 7890.37 818
## .. ... ... ... . ... ... ...
## 41 chr21 33039644-33039688 * | 34.4705 1551.1742 2180
## 42 chr21 33040784-33040798 * | 32.1342 482.0133 2225
## 43 chr21 33040890 * | 30.0925 30.0925 2240
## 44 chr21 33040900-33040901 * | 30.1208 60.2417 2241
## 45 chr21 48019401-48019414 * | 31.1489 436.0850 2243
## indexEnd cluster clusterL
## <integer> <Rle> <Rle>
## 1 565 1 565
## 2 703 2 138
## 3 756 3 366
## 4 817 3 366
## 5 1043 4 765
## .. ... ... ...
## 41 2224 17 45
## 42 2239 18 118
## 43 2240 18 118
## 44 2242 18 118
## 45 2256 19 14
## -------
## seqinfo: 1 sequence from an unspecified genomebpCoverage is the base-level coverage list which can
then be used for plotting.
## Base-level coverage matrices for each of the regions
## Useful for plotting
lapply(regionMat$chr21$bpCoverage[1:2], head, n = 2)## $`1`
## HSB113 HSB123 HSB126 HSB130 HSB135 HSB136 HSB145 HSB153 HSB159 HSB178 HSB92
## 1 93.20 3.32 28.22 5.62 185.17 98.34 5.88 16.71 3.52 15.71 47.40
## 2 124.76 7.25 63.68 11.32 374.85 199.28 10.39 30.53 5.83 29.35 65.04
## HSB97
## 1 36.54
## 2 51.42
##
## $`2`
## HSB113 HSB123 HSB126 HSB130 HSB135 HSB136 HSB145 HSB153 HSB159 HSB178 HSB92
## 566 45.59 7.94 15.92 34.75 141.61 104.21 19.87 38.61 4.97 23.2 13.95
## 567 45.59 7.94 15.92 35.15 141.64 104.30 19.87 38.65 4.97 23.2 13.95
## HSB97
## 566 22.21
## 567 22.21
## Check dimensions. First region is 565 long, second one is 138 bp long.
## The columns match the number of samples (12 in this case).
lapply(regionMat$chr21$bpCoverage[1:2], dim)## $`1`
## [1] 565 12
##
## $`2`
## [1] 138 12The end result of the coverage matrix is shown below. Note that the coverage has been adjusted for read length. Because reads might not fully align inside a given region, the numbers are generally not integers but can be rounded if needed.
## Dimensions of the coverage matrix
dim(regionMat$chr21$coverageMatrix)## [1] 45 12
## Coverage for each region. This matrix can then be used with limma or other pkgs
head(regionMat$chr21$coverageMatrix)## HSB113 HSB123 HSB126 HSB130 HSB135 HSB136
## 1 3653.1093346 277.072106 1397.068687 1106.722895 8987.460401 5570.221054
## 2 333.3740816 99.987237 463.909476 267.354342 1198.713552 1162.313418
## 3 35.3828948 20.153553 30.725394 23.483947 16.786842 17.168947
## 4 42.3398681 29.931579 41.094474 24.724736 32.634080 19.309606
## 5 77.7402631 168.939342 115.059342 171.861974 180.638684 93.503158
## 6 0.7988158 1.770263 1.473421 2.231053 1.697368 1.007895
## HSB145 HSB153 HSB159 HSB178 HSB92 HSB97
## 1 1330.158818 1461.2986829 297.939342 1407.288552 1168.519079 1325.9622371
## 2 257.114210 313.8513139 67.940131 193.695657 127.543553 200.7834228
## 3 22.895921 52.8756585 28.145395 33.127368 23.758816 20.4623685
## 4 33.802632 51.6146040 31.244343 33.576974 29.546183 28.2011836
## 5 90.950526 36.3046051 78.069605 97.151316 100.085790 35.5428946
## 6 1.171316 0.4221053 1.000132 1.139079 1.136447 0.3956579We can then use the coverage matrix and packages such as limma, DESeq2 or edgeR to identify which ERs are differentially expressed. Here we’ll use DESeq2 for which we need some phenotype data.
## Get pheno table
pheno <- subset(brainspanPheno, structure_acronym == "AMY")Now we can identify the DERs using a rounded version of the coverage matrix.
## Loading required package: SummarizedExperiment## Loading required package: MatrixGenerics## Loading required package: matrixStats##
## Attaching package: 'MatrixGenerics'## The following objects are masked from 'package:matrixStats':
##
## colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
## colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
## colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
## colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
## colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
## colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
## colWeightedMeans, colWeightedMedians, colWeightedSds,
## colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
## rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
## rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
## rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
## rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
## rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
## rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
## rowWeightedSds, rowWeightedVars## Loading required package: Biobase## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.##
## Attaching package: 'Biobase'## The following object is masked from 'package:MatrixGenerics':
##
## rowMedians## The following objects are masked from 'package:matrixStats':
##
## anyMissing, rowMedians
## Round matrix
counts <- round(regionMat$chr21$coverageMatrix)
## Round matrix and specify design
dse <- DESeqDataSetFromMatrix(counts, pheno, ~ group + gender)## converting counts to integer mode
## Perform DE analysis
dse <- DESeq(dse, test = "LRT", reduced = ~gender, fitType = "local")## estimating size factors## estimating dispersions## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## fitting model and testing
## Extract results
mcols(regionMat$chr21$regions) <- c(
mcols(regionMat$chr21$regions),
results(dse)
)
## Save info in an object with a shorter name
ers <- regionMat$chr21$regions
ers## GRanges object with 45 ranges and 12 metadata columns:
## seqnames ranges strand | value area indexStart
## <Rle> <IRanges> <Rle> | <numeric> <numeric> <integer>
## 1 chr21 9827018-9827582 * | 313.6717 177224.53 1
## 2 chr21 15457301-15457438 * | 215.0846 29681.68 566
## 3 chr21 20230140-20230192 * | 38.8325 2058.12 704
## 4 chr21 20230445-20230505 * | 41.3245 2520.80 757
## 5 chr21 27253318-27253543 * | 34.9131 7890.37 818
## .. ... ... ... . ... ... ...
## 41 chr21 33039644-33039688 * | 34.4705 1551.1742 2180
## 42 chr21 33040784-33040798 * | 32.1342 482.0133 2225
## 43 chr21 33040890 * | 30.0925 30.0925 2240
## 44 chr21 33040900-33040901 * | 30.1208 60.2417 2241
## 45 chr21 48019401-48019414 * | 31.1489 436.0850 2243
## indexEnd cluster clusterL baseMean log2FoldChange lfcSE stat
## <integer> <Rle> <Rle> <numeric> <numeric> <numeric> <numeric>
## 1 565 1 565 2846.2872 -1.6903182 0.831959 0.215262
## 2 703 2 138 451.5196 -1.1640426 0.757490 0.871126
## 3 756 3 366 29.5781 0.0461488 0.458097 3.132082
## 4 817 3 366 36.0603 -0.1866200 0.390920 2.225708
## 5 1043 4 765 101.6468 -0.1387377 0.320166 3.957987
## .. ... ... ... ... ... ... ...
## 41 2224 17 45 20.782035 -0.642056 0.427661 0.6047814
## 42 2239 18 118 6.410542 -0.634321 0.512262 0.5454039
## 43 2240 18 118 0.129717 -0.859549 3.116540 0.0206273
## 44 2242 18 118 0.702291 -0.628285 2.247378 0.5825105
## 45 2256 19 14 5.293293 -1.694563 1.252290 5.7895910
## pvalue padj
## <numeric> <numeric>
## 1 0.6426743 0.997155
## 2 0.3506436 0.997155
## 3 0.0767657 0.863614
## 4 0.1357305 0.997155
## 5 0.0466495 0.862040
## .. ... ...
## 41 0.4367595 0.997155
## 42 0.4602018 0.997155
## 43 0.8857989 0.997155
## 44 0.4453299 0.997155
## 45 0.0161213 0.725460
## -------
## seqinfo: 1 sequence from an unspecified genomeWe can then compare the DERs against known annotation to see which
DERs overlap known exons, introns, or intergenic regions. A way to
visualize this information is via a Venn diagram which we can create
using vennRegions() from the derfinderPlot
package as shown in Figure @ref(fig:vennRegions).
## Find overlaps between regions and summarized genomic annotation
annoRegs <- annotateRegions(ers, genomicState$fullGenome, verbose = FALSE)
library("derfinderPlot")
venn <- vennRegions(annoRegs,
counts.col = "blue",
main = "Venn diagram using TxDb.Hsapiens.UCSC.hg19.knownGene annotation"
)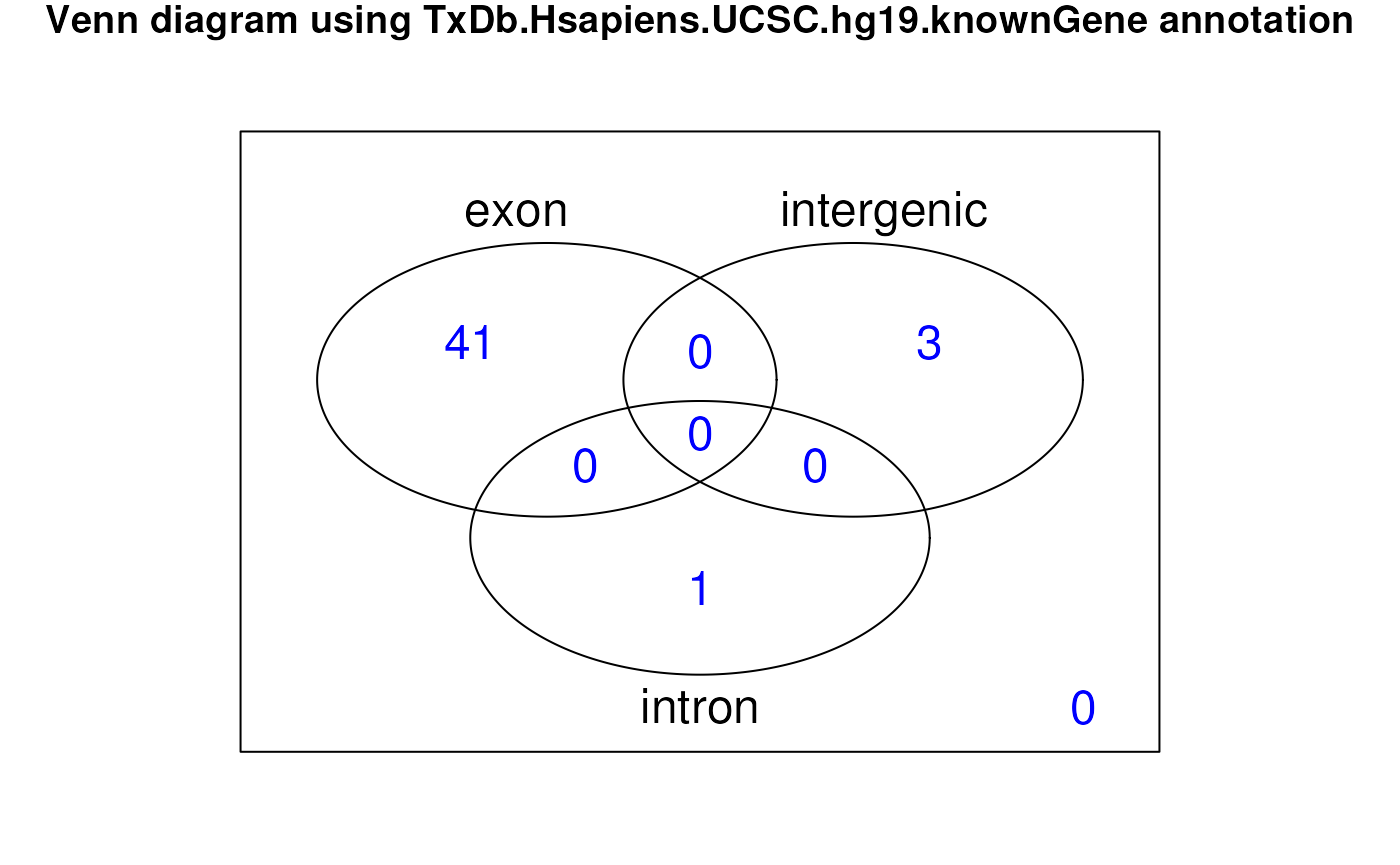
Venn diagram showing ERs by annotation class.
We can also identify the nearest annotated feature. In this case, we’ll look for the nearest known gene from the UCSC hg19 annotation.
## Load database of interest
library("TxDb.Hsapiens.UCSC.hg19.knownGene")## Loading required package: GenomicFeatures## Loading required package: AnnotationDbi
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
seqlevels(txdb) <- "chr21"
## Find nearest feature
library("bumphunter")## Loading required package: foreach## Loading required package: iterators## Loading required package: parallel## Loading required package: locfit## locfit 1.5-9.12 2025-03-05
genes <- annotateTranscripts(txdb)## No annotationPackage supplied. Trying org.Hs.eg.db.## Loading required package: org.Hs.eg.db## Warning in library(package, lib.loc = lib.loc, character.only = TRUE,
## logical.return = TRUE, : there is no package called 'org.Hs.eg.db'## Could not load org.Hs.eg.db. Will continue without annotation## Getting TSS and TSE.## Getting CSS and CSE.## Getting exons.
annotation <- matchGenes(ers, subject = genes)
## Restore seqlevels
txdb <- restoreSeqlevels(txdb)
## View annotation results
head(annotation)## name annotation description region distance subregion
## 1 <NA> <NA> downstream downstream 141014 <NA>
## 2 <NA> <NA> inside intron inside 57537 inside intron
## 3 <NA> <NA> upstream upstream 97502 <NA>
## 4 <NA> <NA> upstream upstream 97807 <NA>
## 5 <NA> <NA> inside exon inside 89287 inside exon
## 6 <NA> <NA> inside exon inside 89283 inside exon
## insideDistance exonnumber nexons UTR strand geneL
## 1 NA NA 5 <NA> - 134665
## 2 -835 2 3 inside transcription region + 74289
## 3 NA NA 3 <NA> - 171451
## 4 NA NA 3 <NA> - 171451
## 5 0 8 8 inside transcription region - 89960
## 6 0 8 8 inside transcription region - 89960
## codingL Geneid subjectHits
## 1 NA 100132288 4
## 2 NA 105377134 664
## 3 NA 101927797 323
## 4 NA 101927797 323
## 5 NA 351 1556
## 6 NA 351 1556
## You can use derfinderPlot::plotOverview() to visualize this informationWe can check the base-level coverage information for some of our DERs. In this example we do so for the first 5 ERs (Figures @ref(fig:firstfive1), @ref(fig:firstfive2), @ref(fig:firstfive3), @ref(fig:firstfive4), @ref(fig:firstfive5)).
## Extract the region coverage
regionCov <- regionMat$chr21$bpCoverage
plotRegionCoverage(
regions = ers, regionCoverage = regionCov,
groupInfo = pheno$group, nearestAnnotation = annotation,
annotatedRegions = annoRegs, whichRegions = seq_len(5), txdb = txdb,
scalefac = 1, ask = FALSE, verbose = FALSE
)## Warning in matrix(c(-pad, -pad, pad, pad), nrow = nrow(d), ncol = 4, byrow =
## TRUE): non-empty data for zero-extent matrix
## Warning in matrix(c(-pad, -pad, pad, pad), nrow = nrow(d), ncol = 4, byrow =
## TRUE): non-empty data for zero-extent matrix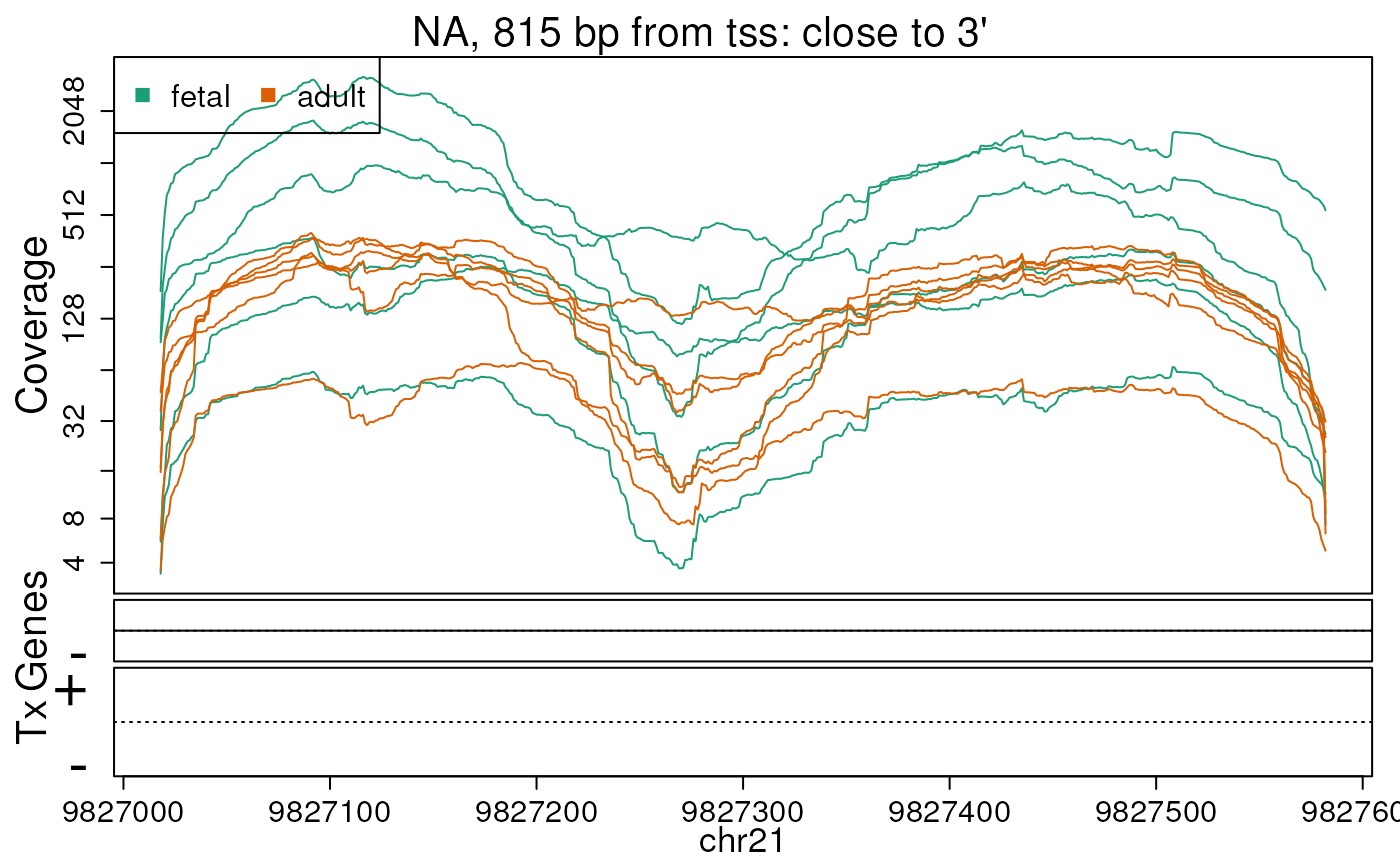
Base-pair resolution plot of differentially expressed region 1.
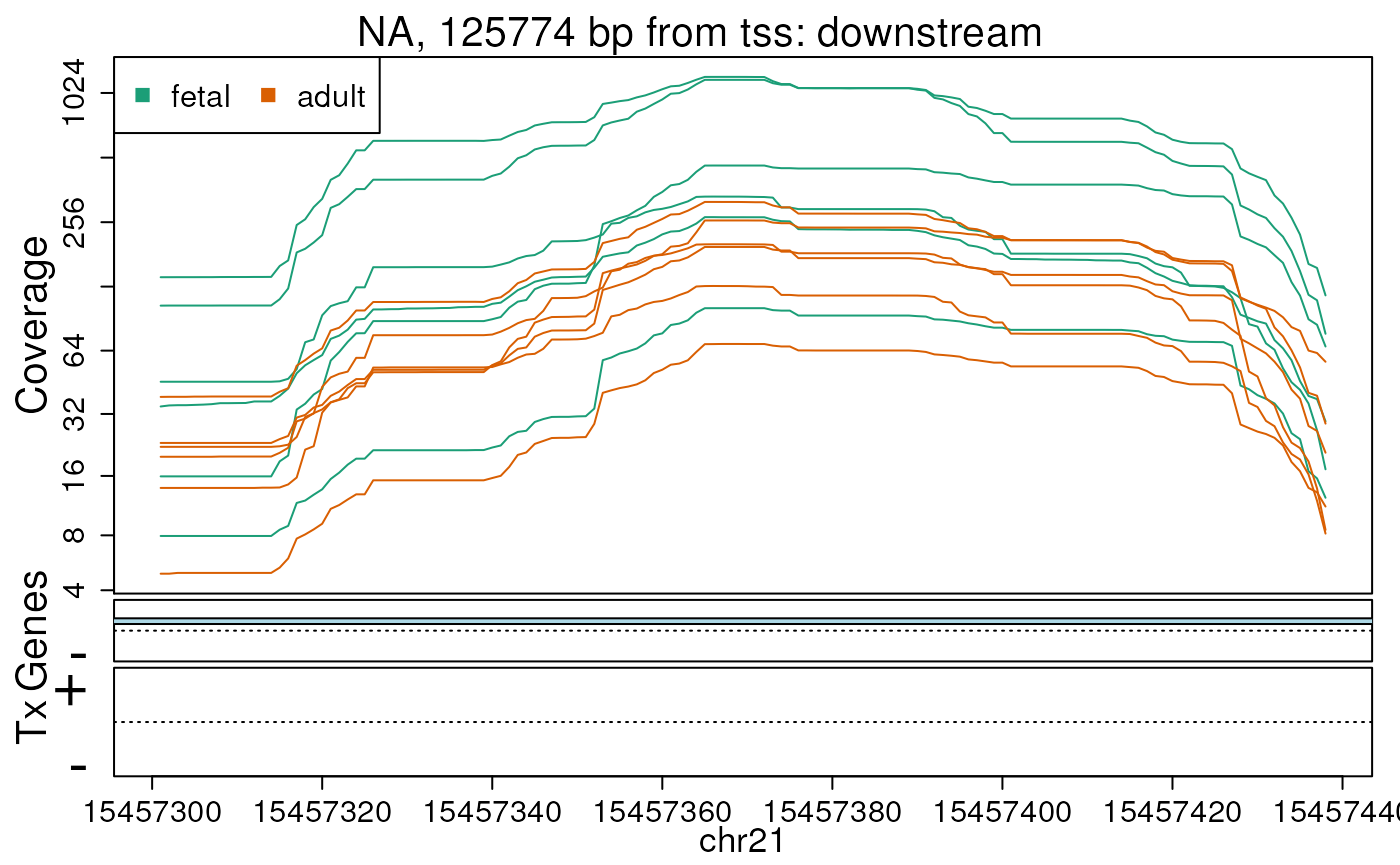
Base-pair resolution plot of differentially expressed region 2.
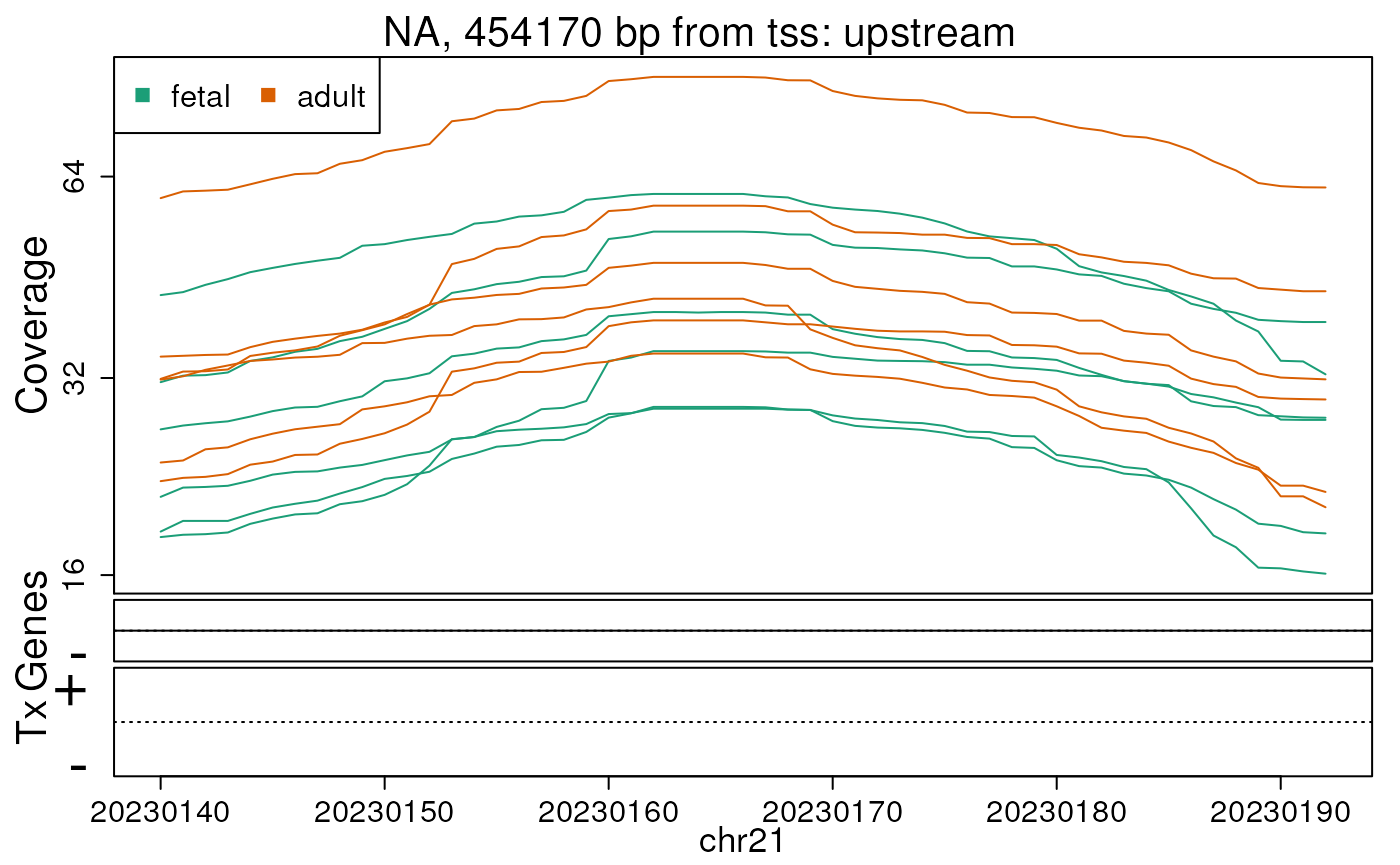
Base-pair resolution plot of differentially expressed region 3.
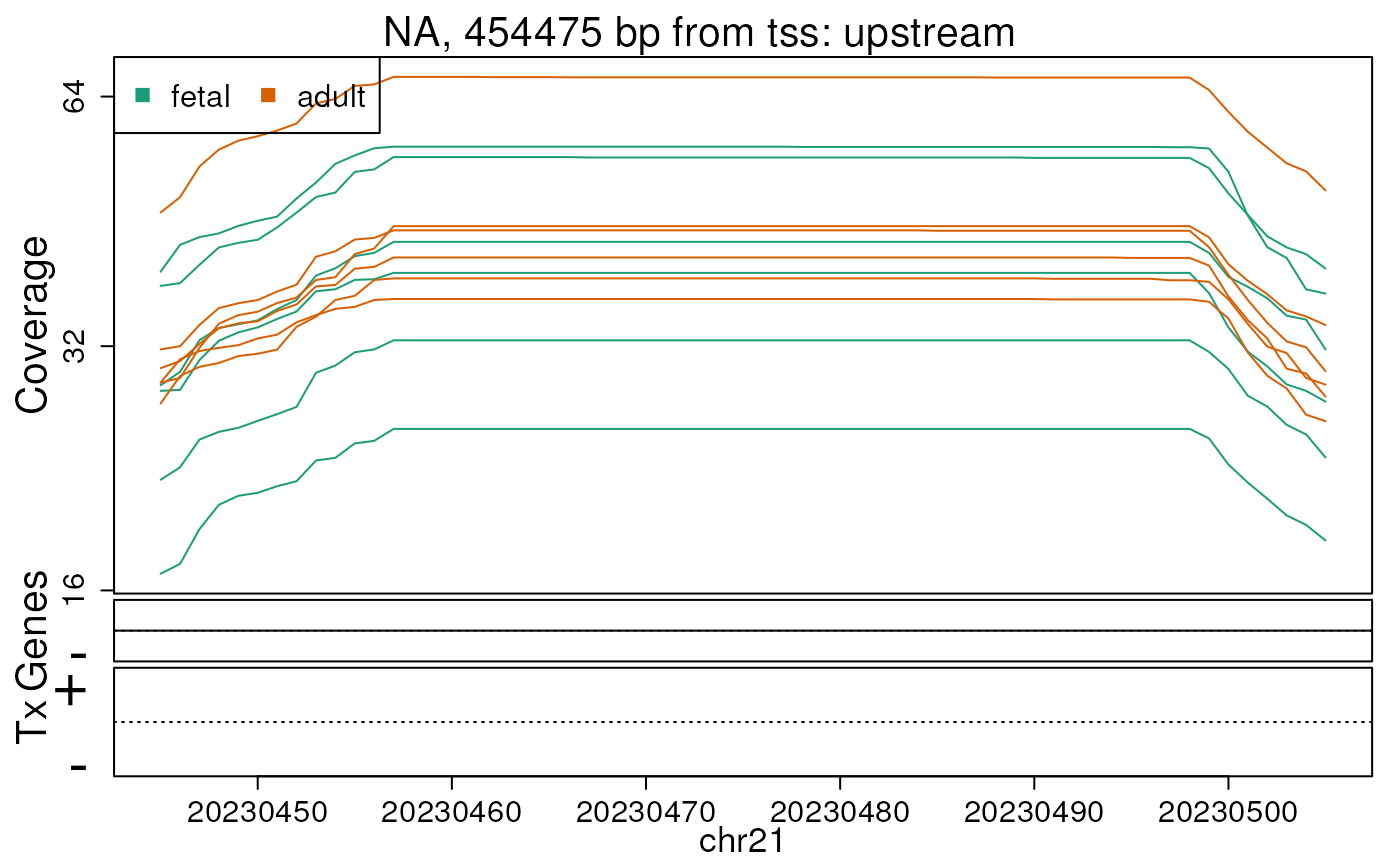
Base-pair resolution plot of differentially expressed region 4.
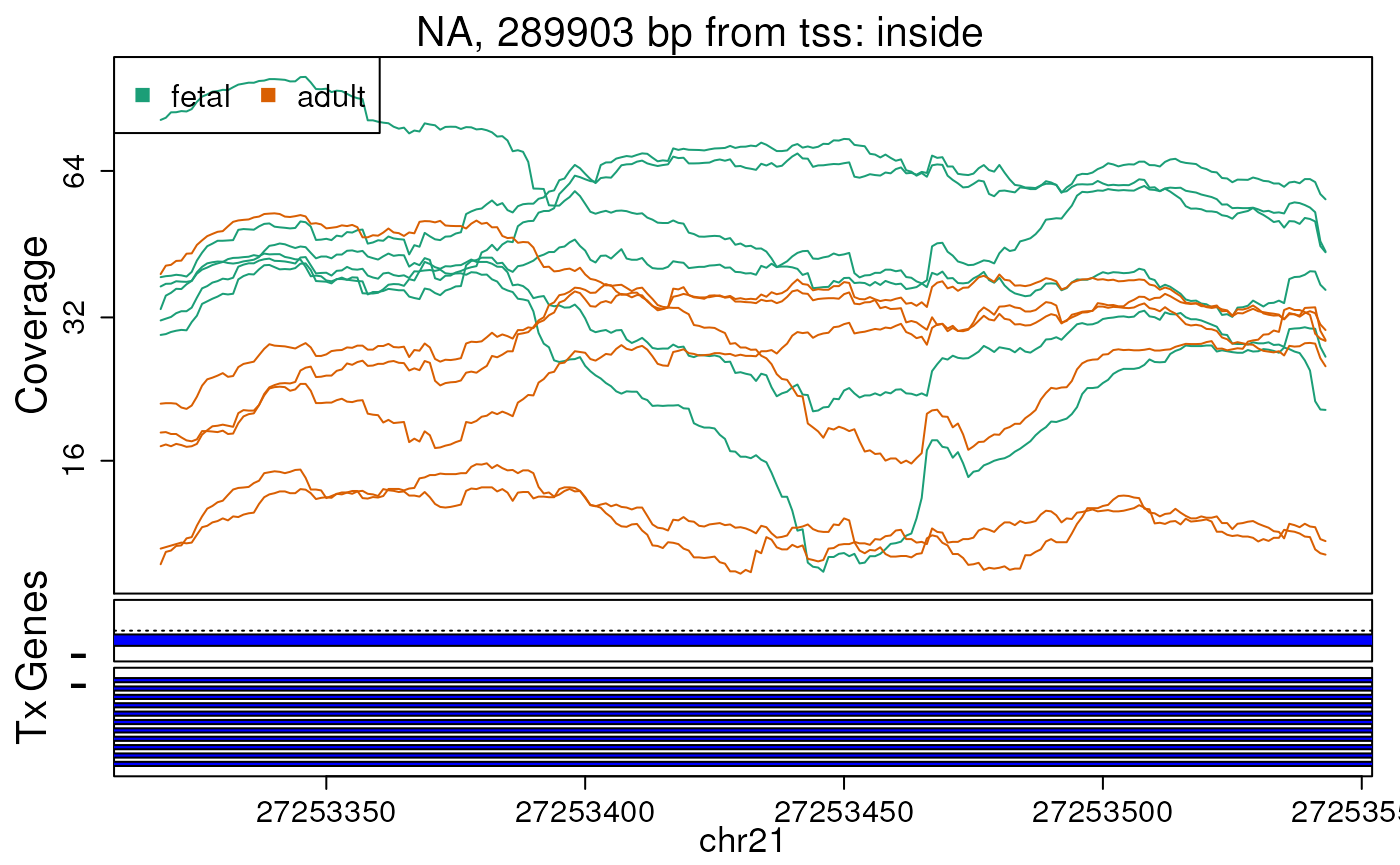
Base-pair resolution plot of differentially expressed region 5.
You can then use the regionReport package to generate interactive HTML reports exploring the results.
If you are interested in using derfinder we recommend checking the derfinder users guide and good luck with your analyses!
Reproducibility
This package was made possible thanks to:
- R (R Core Team, 2026)
- AnnotationDbi (Pagès, Carlson, Falcon, and Li, 2017)
- BiocParallel (Wang, Morgan, Obenchain, Lang, Thompson, and Turaga, 2025)
- bumphunter (Jaffe, Murakami, Lee, Leek, Fallin, Feinberg, and Irizarry, 2012) and (Jaffe, Murakami, Lee, Leek, Fallin, Feinberg, and Irizarry, 2012)
- derfinderHelper (Collado-Torres, Jaffe, and Leek, 2017)
- GenomeInfoDb (Arora, Morgan, Carlson, and Pagès, 2017)
- GenomicAlignments (Lawrence, Huber, Pagès, Aboyoun, Carlson, Gentleman, Morgan, and Carey, 2013)
- GenomicFeatures (Lawrence, Huber, Pagès et al., 2013)
- GenomicFiles (Bioconductor Package Maintainer, Obenchain, Love, Shepherd, and Morgan, 2025)
- GenomicRanges (Lawrence, Huber, Pagès et al., 2013)
- Hmisc (Harrell Jr, 2026)
- IRanges (Lawrence, Huber, Pagès et al., 2013)
- qvalue (Storey, Bass, Dabney, and Robinson, 2025)
- Rsamtools (Morgan, Pagès, Obenchain, and Hayden, 2026)
- rtracklayer (Lawrence, Gentleman, and Carey, 2009)
- S4Vectors (Pagès, Lawrence, and Aboyoun, 2017)
- derfinderData (Collado-Torres, Jaffe, and Leek, 2025)
- sessioninfo (Wickham, Chang, Flight, Müller, and Hester, 2025)
- ggplot2 (Wickham, 2016)
- knitr (Xie, 2014)
- BiocStyle (Oleś, 2025)
- RefManageR (McLean, 2017)
- rmarkdown (Allaire, Xie, Dervieux, McPherson, Luraschi, Ushey, Atkins, Wickham, Cheng, Chang, and Iannone, 2026)
- testthat (Wickham, 2011)
- TxDb.Hsapiens.UCSC.hg19.knownGene (Team and Maintainer, 2025)
Code for creating the vignette
## Create the vignette
library("rmarkdown")
system.time(render("derfinder-quickstart.Rmd", "BiocStyle::html_document"))
## Extract the R code
library("knitr")
knit("derfinder-quickstart.Rmd", tangle = TRUE)Date the vignette was generated.
## [1] "2026-03-31 17:42:10 UTC"Wallclock time spent generating the vignette.
## Time difference of 38.146 secsR session information.
## ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
## setting value
## version R Under development (unstable) (2026-03-28 r89738)
## os Ubuntu 24.04.4 LTS
## system x86_64, linux-gnu
## ui X11
## language en
## collate en_US.UTF-8
## ctype en_US.UTF-8
## tz UTC
## date 2026-03-31
## pandoc 3.9.0.2 @ /usr/bin/ (via rmarkdown)
## quarto 1.8.25 @ /usr/local/bin/quarto
##
## ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
## package * version date (UTC) lib source
## abind 1.4-8 2024-09-12 [1] CRAN (R 4.6.0)
## AnnotationDbi * 1.73.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
## AnnotationFilter 1.35.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
## backports 1.5.0 2024-05-23 [1] CRAN (R 4.6.0)
## base64enc 0.1-6 2026-02-02 [2] CRAN (R 4.7.0)
## bibtex 0.5.2 2026-02-03 [1] CRAN (R 4.6.0)
## Biobase * 2.71.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## BiocGenerics * 0.57.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## BiocIO 1.21.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## BiocManager 1.30.27 2025-11-14 [2] CRAN (R 4.7.0)
## BiocParallel 1.45.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## BiocStyle * 2.39.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## Biostrings 2.79.5 2026-03-06 [1] Bioconductor 3.23 (R 4.7.0)
## biovizBase 1.59.0 2025-11-04 [1] Bioconductor 3.23 (R 4.6.0)
## bit 4.6.0 2025-03-06 [1] CRAN (R 4.6.0)
## bit64 4.6.0-1 2025-01-16 [1] CRAN (R 4.6.0)
## bitops 1.0-9 2024-10-03 [1] CRAN (R 4.6.0)
## blob 1.3.0 2026-01-14 [1] CRAN (R 4.6.0)
## bookdown 0.46 2025-12-05 [1] CRAN (R 4.6.0)
## BSgenome 1.79.1 2025-11-04 [1] Bioconductor 3.23 (R 4.6.0)
## bslib 0.10.0 2026-01-26 [2] CRAN (R 4.7.0)
## bumphunter * 1.53.0 2025-11-04 [1] Bioconductor 3.23 (R 4.6.0)
## cachem 1.1.0 2024-05-16 [2] CRAN (R 4.7.0)
## checkmate 2.3.4 2026-02-03 [1] CRAN (R 4.6.0)
## cigarillo 1.1.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
## cli 3.6.5 2025-04-23 [2] CRAN (R 4.7.0)
## cluster 2.1.8.2 2026-02-05 [3] CRAN (R 4.7.0)
## codetools 0.2-20 2024-03-31 [3] CRAN (R 4.7.0)
## colorspace 2.1-2 2025-09-22 [1] CRAN (R 4.6.0)
## crayon 1.5.3 2024-06-20 [2] CRAN (R 4.7.0)
## curl 7.0.0 2025-08-19 [2] CRAN (R 4.7.0)
## data.table 1.18.2.1 2026-01-27 [1] CRAN (R 4.6.0)
## DBI 1.3.0 2026-02-25 [1] CRAN (R 4.7.0)
## DelayedArray 0.37.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
## derfinder * 1.45.0 2026-03-31 [1] Bioconductor
## derfinderData * 2.29.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## derfinderHelper 1.45.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
## derfinderPlot * 1.45.0 2025-11-04 [1] Bioconductor 3.23 (R 4.6.0)
## desc 1.4.3 2023-12-10 [2] CRAN (R 4.7.0)
## DESeq2 * 1.51.7 2026-03-13 [1] Bioconductor 3.23 (R 4.7.0)
## dichromat 2.0-0.1 2022-05-02 [1] CRAN (R 4.6.0)
## digest 0.6.39 2025-11-19 [2] CRAN (R 4.7.0)
## doRNG 1.8.6.3 2026-02-05 [1] CRAN (R 4.6.0)
## dplyr 1.2.0 2026-02-03 [1] CRAN (R 4.6.0)
## ensembldb 2.35.0 2025-11-04 [1] Bioconductor 3.23 (R 4.6.0)
## evaluate 1.0.5 2025-08-27 [2] CRAN (R 4.7.0)
## farver 2.1.2 2024-05-13 [1] CRAN (R 4.6.0)
## fastmap 1.2.0 2024-05-15 [2] CRAN (R 4.7.0)
## foreach * 1.5.2 2022-02-02 [1] CRAN (R 4.6.0)
## foreign 0.8-91 2026-01-29 [3] CRAN (R 4.7.0)
## Formula 1.2-5 2023-02-24 [1] CRAN (R 4.6.0)
## fs 2.0.1 2026-03-24 [2] CRAN (R 4.7.0)
## generics * 0.1.4 2025-05-09 [1] CRAN (R 4.6.0)
## GenomeInfoDb 1.47.2 2025-12-04 [1] Bioconductor 3.23 (R 4.6.0)
## GenomicAlignments 1.47.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
## GenomicFeatures * 1.63.1 2025-11-04 [1] Bioconductor 3.23 (R 4.6.0)
## GenomicFiles 1.47.0 2025-11-04 [1] Bioconductor 3.23 (R 4.6.0)
## GenomicRanges * 1.63.1 2025-12-08 [1] Bioconductor 3.23 (R 4.6.0)
## ggbio 1.59.0 2025-11-04 [1] Bioconductor 3.23 (R 4.6.0)
## ggplot2 4.0.2 2026-02-03 [1] CRAN (R 4.6.0)
## glue 1.8.0 2024-09-30 [2] CRAN (R 4.7.0)
## graph 1.89.1 2025-12-02 [1] Bioconductor 3.23 (R 4.6.0)
## gridExtra 2.3 2017-09-09 [1] CRAN (R 4.6.0)
## gtable 0.3.6 2024-10-25 [1] CRAN (R 4.6.0)
## Hmisc 5.2-5 2026-01-09 [1] CRAN (R 4.6.0)
## htmlTable 2.4.3 2024-07-21 [1] CRAN (R 4.6.0)
## htmltools 0.5.9 2025-12-04 [2] CRAN (R 4.7.0)
## htmlwidgets 1.6.4 2023-12-06 [2] CRAN (R 4.7.0)
## httr 1.4.8 2026-02-13 [1] CRAN (R 4.7.0)
## IRanges * 2.45.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
## iterators * 1.0.14 2022-02-05 [1] CRAN (R 4.6.0)
## jquerylib 0.1.4 2021-04-26 [2] CRAN (R 4.7.0)
## jsonlite 2.0.0 2025-03-27 [2] CRAN (R 4.7.0)
## KEGGREST 1.51.1 2025-11-17 [1] Bioconductor 3.23 (R 4.6.0)
## knitr 1.51 2025-12-20 [2] CRAN (R 4.7.0)
## lattice 0.22-9 2026-02-09 [3] CRAN (R 4.7.0)
## lazyeval 0.2.2 2019-03-15 [1] CRAN (R 4.6.0)
## lifecycle 1.0.5 2026-01-08 [2] CRAN (R 4.7.0)
## limma 3.67.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## locfit * 1.5-9.12 2025-03-05 [1] CRAN (R 4.6.0)
## lubridate 1.9.5 2026-02-04 [1] CRAN (R 4.6.0)
## magrittr 2.0.4 2025-09-12 [2] CRAN (R 4.7.0)
## Matrix 1.7-5 2026-03-21 [3] CRAN (R 4.7.0)
## MatrixGenerics * 1.23.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## matrixStats * 1.5.0 2025-01-07 [1] CRAN (R 4.6.0)
## memoise 2.0.1 2021-11-26 [2] CRAN (R 4.7.0)
## nnet 7.3-20 2025-01-01 [3] CRAN (R 4.7.0)
## OrganismDbi 1.53.2 2025-11-04 [1] Bioconductor 3.23 (R 4.6.0)
## otel 0.2.0 2025-08-29 [2] CRAN (R 4.7.0)
## pillar 1.11.1 2025-09-17 [2] CRAN (R 4.7.0)
## pkgconfig 2.0.3 2019-09-22 [2] CRAN (R 4.7.0)
## pkgdown 2.2.0.9000 2026-03-31 [1] Github (r-lib/pkgdown@a6abe43)
## plyr 1.8.9 2023-10-02 [1] CRAN (R 4.6.0)
## png 0.1-9 2026-03-15 [1] CRAN (R 4.7.0)
## ProtGenerics 1.43.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## qvalue 2.43.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## R6 2.6.1 2025-02-15 [2] CRAN (R 4.7.0)
## ragg 1.5.2 2026-03-23 [2] CRAN (R 4.7.0)
## RBGL 1.87.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## RColorBrewer 1.1-3 2022-04-03 [1] CRAN (R 4.6.0)
## Rcpp 1.1.1 2026-01-10 [2] CRAN (R 4.7.0)
## RCurl 1.98-1.18 2026-03-21 [1] CRAN (R 4.7.0)
## RefManageR * 1.4.0 2022-09-30 [1] CRAN (R 4.6.0)
## reshape2 1.4.5 2025-11-12 [1] CRAN (R 4.6.0)
## restfulr 0.0.16 2025-06-27 [1] CRAN (R 4.6.0)
## rjson 0.2.23 2024-09-16 [1] CRAN (R 4.6.0)
## rlang 1.1.7 2026-01-09 [2] CRAN (R 4.7.0)
## rmarkdown 2.31 2026-03-26 [2] CRAN (R 4.7.0)
## rngtools 1.5.2 2021-09-20 [1] CRAN (R 4.6.0)
## rpart 4.1.27 2026-03-27 [3] CRAN (R 4.7.0)
## Rsamtools 2.27.1 2026-03-08 [1] Bioconductor 3.23 (R 4.7.0)
## RSQLite 2.4.6 2026-02-06 [1] CRAN (R 4.6.0)
## rstudioapi 0.18.0 2026-01-16 [2] CRAN (R 4.7.0)
## rtracklayer 1.71.3 2025-12-14 [1] Bioconductor 3.23 (R 4.6.0)
## S4Arrays 1.11.1 2025-11-25 [1] Bioconductor 3.23 (R 4.6.0)
## S4Vectors * 0.49.0 2025-10-30 [1] Bioconductor 3.23 (R 4.6.0)
## S7 0.2.1 2025-11-14 [1] CRAN (R 4.6.0)
## sass 0.4.10 2025-04-11 [2] CRAN (R 4.7.0)
## scales 1.4.0 2025-04-24 [1] CRAN (R 4.6.0)
## Seqinfo * 1.1.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
## sessioninfo * 1.2.3 2025-02-05 [2] CRAN (R 4.7.0)
## SparseArray 1.11.12 2026-03-30 [1] Bioconductor 3.23 (R 4.7.0)
## statmod 1.5.1 2025-10-09 [1] CRAN (R 4.6.0)
## stringi 1.8.7 2025-03-27 [2] CRAN (R 4.7.0)
## stringr 1.6.0 2025-11-04 [2] CRAN (R 4.7.0)
## SummarizedExperiment * 1.41.1 2026-02-06 [1] Bioconductor 3.23 (R 4.6.0)
## systemfonts 1.3.2 2026-03-05 [2] CRAN (R 4.7.0)
## textshaping 1.0.5 2026-03-06 [2] CRAN (R 4.7.0)
## tibble 3.3.1 2026-01-11 [2] CRAN (R 4.7.0)
## tidyselect 1.2.1 2024-03-11 [1] CRAN (R 4.6.0)
## timechange 0.4.0 2026-01-29 [1] CRAN (R 4.6.0)
## TxDb.Hsapiens.UCSC.hg19.knownGene * 3.22.1 2026-02-11 [1] Bioconductor
## UCSC.utils 1.7.1 2025-12-09 [1] Bioconductor 3.23 (R 4.6.0)
## VariantAnnotation 1.57.1 2025-12-18 [1] Bioconductor 3.23 (R 4.6.0)
## vctrs 0.7.2 2026-03-21 [2] CRAN (R 4.7.0)
## xfun 0.57 2026-03-20 [2] CRAN (R 4.7.0)
## XML 3.99-0.23 2026-03-20 [1] CRAN (R 4.7.0)
## xml2 1.5.2 2026-01-17 [2] CRAN (R 4.7.0)
## XVector 0.51.0 2025-10-31 [1] Bioconductor 3.23 (R 4.6.0)
## yaml 2.3.12 2025-12-10 [2] CRAN (R 4.7.0)
##
## [1] /__w/_temp/Library
## [2] /usr/local/lib/R/site-library
## [3] /usr/local/lib/R/library
## * ── Packages attached to the search path.
##
## ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────Bibliography
This vignette was generated using BiocStyle (Oleś, 2025) with knitr (Xie, 2014) and rmarkdown (Allaire, Xie, Dervieux et al., 2026) running behind the scenes.
Citations made with RefManageR (McLean, 2017).
[1] J. Allaire, Y. Xie, C. Dervieux, et al. rmarkdown: Dynamic Documents for R. R package version 2.31. 2026. URL: https://github.com/rstudio/rmarkdown.
[2] S. Arora, M. Morgan, M. Carlson, et al. GenomeInfoDb: Utilities for manipulating chromosome and other ‘seqname’ identifiers. 2017. DOI: 10.18129/B9.bioc.GenomeInfoDb.
[3] Bioconductor Package Maintainer, V. Obenchain, M. Love, et al. GenomicFiles: Distributed computing by file or by range. R package version 1.47.0. 2025. DOI: 10.18129/B9.bioc.GenomicFiles. URL: https://bioconductor.org/packages/GenomicFiles.
[4] BrainSpan. “Atlas of the Developing Human Brain [Internet]. Funded by ARRA Awards 1RC2MH089921-01, 1RC2MH090047-01, and 1RC2MH089929-01.” 2011. URL: http://www.brainspan.org/.
[5] L. Collado-Torres, A. E. Jaffe, and J. T. Leek. derfinderHelper: derfinder helper package. https://github.com/leekgroup/derfinderHelper - R package version 1.45.0. 2017. DOI: 10.18129/B9.bioc.derfinderHelper. URL: http://www.bioconductor.org/packages/derfinderHelper.
[6] L. Collado-Torres, A. Jaffe, and J. Leek. derfinderData: Processed BigWigs from BrainSpan for examples. R package version 2.29.0. 2025. DOI: 10.18129/B9.bioc.derfinderData. URL: https://bioconductor.org/packages/derfinderData.
[7] A. C. Frazee, S. Sabunciyan, K. D. Hansen, et al. “Differential expression analysis of RNA-seq data at single-base resolution”. In: Biostatistics 15 (3) (2014), pp. 413-426. DOI: 10.1093/biostatistics/kxt053. URL: http://biostatistics.oxfordjournals.org/content/15/3/413.long.
[8] F. Harrell Jr. Hmisc: Harrell Miscellaneous. R package version 5.2-5. 2026. DOI: 10.32614/CRAN.package.Hmisc. URL: https://CRAN.R-project.org/package=Hmisc.
[9] A. E. Jaffe, P. Murakami, H. Lee, et al. “Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies”. In: International journal of epidemiology 41.1 (2012), pp. 200–209. DOI: 10.1093/ije/dyr238.
[10] A. E. Jaffe, P. Murakami, H. Lee, et al. “Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies”. In: International Journal of Epidemiology (2012).
[11] M. Lawrence, R. Gentleman, and V. Carey. “rtracklayer: an R package for interfacing with genome browsers”. In: Bioinformatics 25 (2009), pp. 1841-1842. DOI: 10.1093/bioinformatics/btp328. URL: http://bioinformatics.oxfordjournals.org/content/25/14/1841.abstract.
[12] M. Lawrence, W. Huber, H. Pagès, et al. “Software for Computing and Annotating Genomic Ranges”. In: PLoS Computational Biology 9 (8 2013). DOI: 10.1371/journal.pcbi.1003118. URL: http://www.ploscompbiol.org/article/info%3Adoi%2F10.1371%2Fjournal.pcbi.1003118}.
[13] M. W. McLean. “RefManageR: Import and Manage BibTeX and BibLaTeX References in R”. In: The Journal of Open Source Software (2017). DOI: 10.21105/joss.00338.
[14] M. Morgan, H. Pagès, V. Obenchain, et al. Rsamtools: Binary alignment (BAM), FASTA, variant call (BCF), and tabix file import. R package version 2.27.1. 2026. DOI: 10.18129/B9.bioc.Rsamtools. URL: https://bioconductor.org/packages/Rsamtools.
[15] A. Oleś. BiocStyle: Standard styles for vignettes and other Bioconductor documents. R package version 2.39.0. 2025. DOI: 10.18129/B9.bioc.BiocStyle. URL: https://bioconductor.org/packages/BiocStyle.
[16] H. Pagès, M. Carlson, S. Falcon, et al. AnnotationDbi: Annotation Database Interface. 2017. DOI: 10.18129/B9.bioc.AnnotationDbi.
[17] H. Pagès, M. Lawrence, and P. Aboyoun. S4Vectors: S4 implementation of vector-like and list-like objects. 2017. DOI: 10.18129/B9.bioc.S4Vectors.
[18] R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing (ROR: <https://ror.org/05qewa988>;). Vienna, Austria, 2026. DOI: 10.32614/R.manuals. URL: https://www.R-project.org/.
[19] J. D. Storey, A. J. Bass, A. Dabney, et al. qvalue: Q-value estimation for false discovery rate control. R package version 2.43.0. 2025. DOI: 10.18129/B9.bioc.qvalue. URL: https://bioconductor.org/packages/qvalue.
[20] B. C. Team and B. P. Maintainer. TxDb.Hsapiens.UCSC.hg19.knownGene: Annotation package for TxDb object(s). R package version 3.22.1. 2025.
[21] J. Wang, M. Morgan, V. Obenchain, et al. BiocParallel: Bioconductor facilities for parallel evaluation. R package version 1.45.0. 2025. DOI: 10.18129/B9.bioc.BiocParallel. URL: https://bioconductor.org/packages/BiocParallel.
[22] H. Wickham. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York, 2016. ISBN: 978-3-319-24277-4. URL: https://ggplot2.tidyverse.org.
[23] H. Wickham. “testthat: Get Started with Testing”. In: The R Journal 3 (2011), pp. 5–10. URL: https://journal.r-project.org/articles/RJ-2011-002/.
[24] H. Wickham, W. Chang, R. Flight, et al. sessioninfo: R Session Information. R package version 1.2.3. 2025. DOI: 10.32614/CRAN.package.sessioninfo. URL: https://CRAN.R-project.org/package=sessioninfo.
[25] Y. Xie. “knitr: A Comprehensive Tool for Reproducible Research in R”. In: Implementing Reproducible Computational Research. Ed. by V. Stodden, F. Leisch and R. D. Peng. ISBN 978-1466561595. Chapman and Hall/CRC, 2014.