6 Interpreting model coefficients with ExploreModelMatrix
Instructor: Leo
6.1 Model objects in R
- Linear regression review https://lcolladotor.github.io/bioc_team_ds/helping-others.html#linear-regression-example
- With R, we use the
model.matrix()to build regression models using theY ~ X1 + X2formula syntax as exemplified below.
## ?model.matrix
mat <- with(trees, model.matrix(log(Volume) ~ log(Height) + log(Girth)))
mat
#> (Intercept) log(Height) log(Girth)
#> 1 1 4.248495 2.116256
#> 2 1 4.174387 2.151762
#> 3 1 4.143135 2.174752
#> 4 1 4.276666 2.351375
#> 5 1 4.394449 2.370244
#> 6 1 4.418841 2.379546
#> 7 1 4.189655 2.397895
#> 8 1 4.317488 2.397895
#> 9 1 4.382027 2.406945
#> 10 1 4.317488 2.415914
#> 11 1 4.369448 2.424803
#> 12 1 4.330733 2.433613
#> 13 1 4.330733 2.433613
#> 14 1 4.234107 2.459589
#> 15 1 4.317488 2.484907
#> 16 1 4.304065 2.557227
#> [ reached getOption("max.print") -- omitted 15 rows ]
#> attr(,"assign")
#> [1] 0 1 2- How do we interpret the columns of our model matrix
mat?
summary(lm(log(Volume) ~ log(Height) + log(Girth), data = trees))
#>
#> Call:
#> lm(formula = log(Volume) ~ log(Height) + log(Girth), data = trees)
#>
#> Residuals:
#> Min 1Q Median 3Q Max
#> -0.168561 -0.048488 0.002431 0.063637 0.129223
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> (Intercept) -6.63162 0.79979 -8.292 5.06e-09 ***
#> log(Height) 1.11712 0.20444 5.464 7.81e-06 ***
#> log(Girth) 1.98265 0.07501 26.432 < 2e-16 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Residual standard error: 0.08139 on 28 degrees of freedom
#> Multiple R-squared: 0.9777, Adjusted R-squared: 0.9761
#> F-statistic: 613.2 on 2 and 28 DF, p-value: < 2.2e-166.2 ExploreModelMatrix
- It’s a Bioconductor package which is useful to understand statistical models we use in differential expression analyses. It is interactive and helps us by creating some visual aids.
- For more details, check their paper https://doi.org/10.12688/f1000research.24187.2.
- We’ll go over the examples they provide at http://www.bioconductor.org/packages/release/bioc/vignettes/ExploreModelMatrix/inst/doc/ExploreModelMatrix.html
6.3 Example 1
## Load ExploreModelMatrix
library("ExploreModelMatrix")
## Example data
(sampleData <- data.frame(
genotype = rep(c("A", "B"), each = 4),
treatment = rep(c("ctrl", "trt"), 4)
))
#> genotype treatment
#> 1 A ctrl
#> 2 A trt
#> 3 A ctrl
#> 4 A trt
#> 5 B ctrl
#> 6 B trt
#> 7 B ctrl
#> 8 B trt
## Let's make the visual aids provided by ExploreModelMatrix
vd <- ExploreModelMatrix::VisualizeDesign(
sampleData = sampleData,
designFormula = ~ genotype + treatment,
textSizeFitted = 4
)
## Now lets plot these images
cowplot::plot_grid(plotlist = vd$plotlist)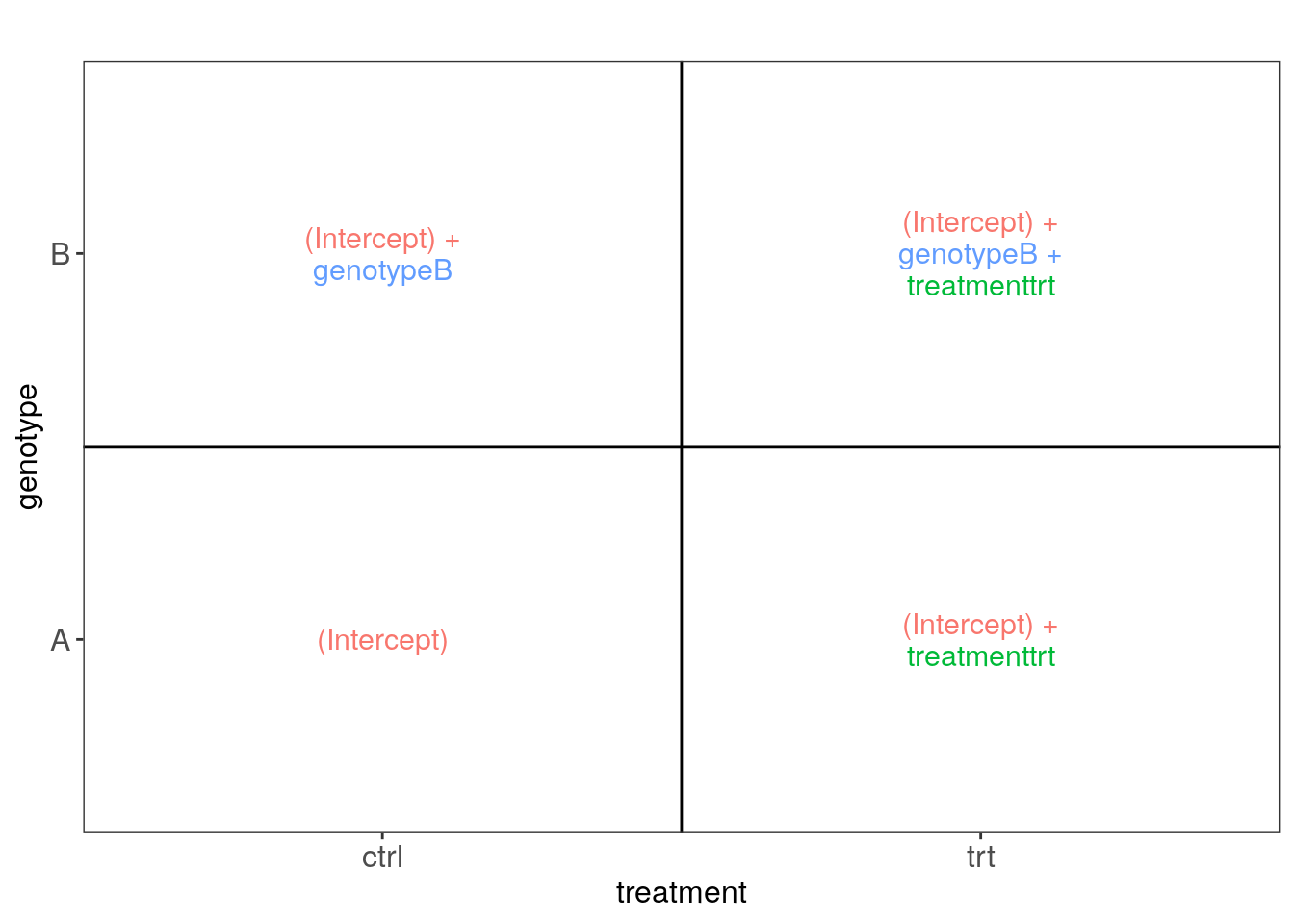
Interactively, we can run the following code:
6.6 Exercise
Exercise 1:
Interpret ResponseResistant.Treatmentpre from the second example. It could be useful to take a screenshot and to draw some annotations on it.
Exercise 2:
Whis is the 0 important at the beginning of the formula in the third example?
6.7 To learn more
A guide to creating design matrices for gene expression experiments:
- http://bioconductor.org/packages/release/workflows/vignettes/RNAseq123/inst/doc/designmatrices.html
- https://f1000research.com/articles/9-1444
“Model matrix not full rank”
6.8 Community
Some of the ExploreModelMatrix authors:
- https://bsky.app/profile/csoneson.bsky.social
- https://twitter.com/FedeBioinfo
- https://twitter.com/mikelove
Some of the edgeR and limma authors: