1 SummarizedExperiment overview
Instructor: Leo
1.1 Overview
The SummarizedExperiment class is used to store experimental results in the form of matrixes. Objects of this class include observations (features) of the samples, as well as additional metadata. Usually, this type of object is automatically generated as the output of other software (ie. SPEAQeasy), but you can also build them.
One of the main characteristics of SummarizedExperiment is that it allows you to handle you data in a “coordinated” way. For example, if you want to subset your data, with SummarizedExperiment you can do so without worrying about keeping your assays and metadata synched.
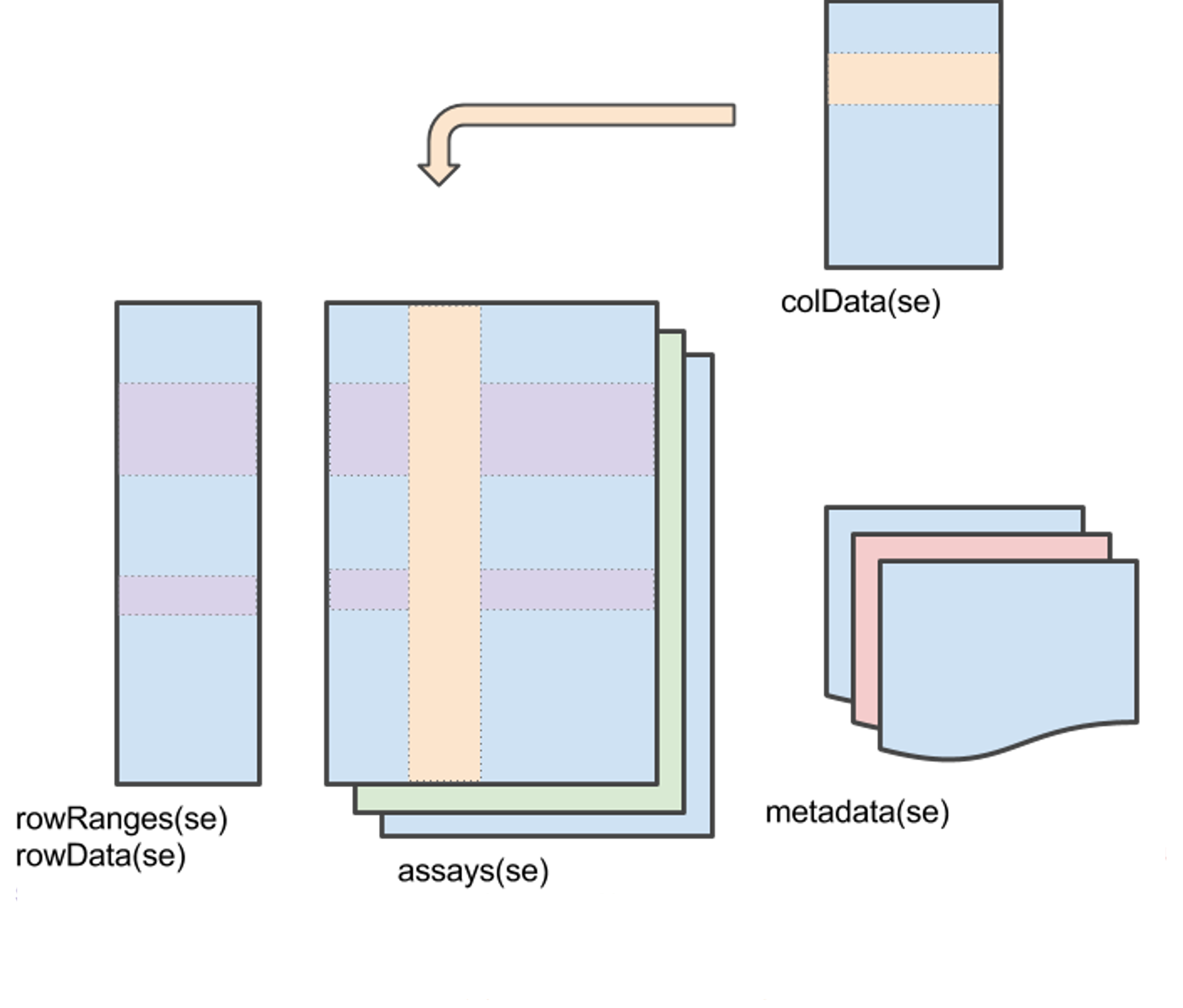
1.2 Exercises
We are gonna use the sample data set from the airway library.
Exercise 1:
a) How many genes do we have in this object? And samples?
b) How many samples come from donors treated (trt) with dexamethasone (dex)?
## For a) you could only print the summary of the object but since the idea is
## to understand how to explore the object find other function that gives
## you the answer.
se
#> class: RangedSummarizedExperiment
#> dim: 63677 8
#> metadata(1): ''
#> assays(1): counts
#> rownames(63677): ENSG00000000003 ENSG00000000005 ... ENSG00000273492 ENSG00000273493
#> rowData names(10): gene_id gene_name ... seq_coord_system symbol
#> colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
#> colData names(9): SampleName cell ... Sample BioSample
## Same thing for b, you could just print the colData and count the samples,
## but this is not efficient when our data consists in hundreds of samples.
## Find the answer using other tools.
colData(se)
#> DataFrame with 8 rows and 9 columns
#> SampleName cell dex albut Run avgLength Experiment Sample BioSample
#> <factor> <factor> <factor> <factor> <factor> <integer> <factor> <factor> <factor>
#> SRR1039508 GSM1275862 N61311 untrt untrt SRR1039508 126 SRX384345 SRS508568 SAMN02422669
#> SRR1039509 GSM1275863 N61311 trt untrt SRR1039509 126 SRX384346 SRS508567 SAMN02422675
#> SRR1039512 GSM1275866 N052611 untrt untrt SRR1039512 126 SRX384349 SRS508571 SAMN02422678
#> SRR1039513 GSM1275867 N052611 trt untrt SRR1039513 87 SRX384350 SRS508572 SAMN02422670
#> [ reached getOption("max.print") -- omitted 4 rows ]Exercise 2: Add another assay that has the log10 of your original counts
## In our object, if you look at the part that says assays, we can see that
## at the moment we only have one with the name "counts".
se
#> class: RangedSummarizedExperiment
#> dim: 63677 8
#> metadata(1): ''
#> assays(1): counts
#> rownames(63677): ENSG00000000003 ENSG00000000005 ... ENSG00000273492 ENSG00000273493
#> rowData names(10): gene_id gene_name ... seq_coord_system symbol
#> colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
#> colData names(9): SampleName cell ... Sample BioSample
## To see the data that's stored in that assay you can do either one of the
## next commands.
assay(se)
#> SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517 SRR1039520 SRR1039521
#> ENSG00000000003 679 448 873 408 1138 1047 770 572
#> ENSG00000000005 0 0 0 0 0 0 0 0
#> ENSG00000000419 467 515 621 365 587 799 417 508
#> ENSG00000000457 260 211 263 164 245 331 233 229
#> ENSG00000000460 60 55 40 35 78 63 76 60
#> ENSG00000000938 0 0 2 0 1 0 0 0
#> [ reached getOption("max.print") -- omitted 63671 rows ]assays(se)$counts
#> SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517 SRR1039520 SRR1039521
#> ENSG00000000003 679 448 873 408 1138 1047 770 572
#> ENSG00000000005 0 0 0 0 0 0 0 0
#> ENSG00000000419 467 515 621 365 587 799 417 508
#> ENSG00000000457 260 211 263 164 245 331 233 229
#> ENSG00000000460 60 55 40 35 78 63 76 60
#> ENSG00000000938 0 0 2 0 1 0 0 0
#> [ reached getOption("max.print") -- omitted 63671 rows ]
## Note that assay() does not support $ operator
# assay(se)$counts
## We would have to do:
assay(se, 1)
#> SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517 SRR1039520 SRR1039521
#> ENSG00000000003 679 448 873 408 1138 1047 770 572
#> ENSG00000000005 0 0 0 0 0 0 0 0
#> ENSG00000000419 467 515 621 365 587 799 417 508
#> ENSG00000000457 260 211 263 164 245 331 233 229
#> ENSG00000000460 60 55 40 35 78 63 76 60
#> ENSG00000000938 0 0 2 0 1 0 0 0
#> [ reached getOption("max.print") -- omitted 63671 rows ]assay(se, "counts")
#> SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517 SRR1039520 SRR1039521
#> ENSG00000000003 679 448 873 408 1138 1047 770 572
#> ENSG00000000005 0 0 0 0 0 0 0 0
#> ENSG00000000419 467 515 621 365 587 799 417 508
#> ENSG00000000457 260 211 263 164 245 331 233 229
#> ENSG00000000460 60 55 40 35 78 63 76 60
#> ENSG00000000938 0 0 2 0 1 0 0 0
#> [ reached getOption("max.print") -- omitted 63671 rows ]
## If you use assays() without specifying the element you want to see it
## shows you the length of the list and the name of each element.
assays(se)
#> List of length 1
#> names(1): counts
## Which can also be use to change the name of the assays
assayNames(se)[1] <- "foo"
assayNames(se)
#> [1] "foo"Exercise 3: Explore the metadata and add a new column that has the library size of each sample.
1.3 Solutions
Solution 1:
## For b),
colData(se)[colData(se)$dex == "trt", ]
#> DataFrame with 4 rows and 9 columns
#> SampleName cell dex albut Run avgLength Experiment Sample BioSample
#> <factor> <factor> <factor> <factor> <factor> <integer> <factor> <factor> <factor>
#> SRR1039509 GSM1275863 N61311 trt untrt SRR1039509 126 SRX384346 SRS508567 SAMN02422675
#> SRR1039513 GSM1275867 N052611 trt untrt SRR1039513 87 SRX384350 SRS508572 SAMN02422670
#> SRR1039517 GSM1275871 N080611 trt untrt SRR1039517 126 SRX384354 SRS508576 SAMN02422673
#> SRR1039521 GSM1275875 N061011 trt untrt SRR1039521 98 SRX384358 SRS508580 SAMN02422677colData(se)[se$dex == "trt", ]
#> DataFrame with 4 rows and 9 columns
#> SampleName cell dex albut Run avgLength Experiment Sample BioSample
#> <factor> <factor> <factor> <factor> <factor> <integer> <factor> <factor> <factor>
#> SRR1039509 GSM1275863 N61311 trt untrt SRR1039509 126 SRX384346 SRS508567 SAMN02422675
#> SRR1039513 GSM1275867 N052611 trt untrt SRR1039513 87 SRX384350 SRS508572 SAMN02422670
#> SRR1039517 GSM1275871 N080611 trt untrt SRR1039517 126 SRX384354 SRS508576 SAMN02422673
#> SRR1039521 GSM1275875 N061011 trt untrt SRR1039521 98 SRX384358 SRS508580 SAMN02422677Solution 2:
## There are multiple ways to do it
assay(se, "logcounts") <- log10(assay(se, "counts"))
assays(se)$logcounts_v2 <- log10(assays(se)$counts)Solution 3: